
Sarcopenia in the setting of nonalcoholic fatty liver


Abstract
Nonalcoholic fatty liver is a worldwide common problem with more prevalence in non-Asian populations that is closely correlated with the muscle-related disorder sarcopenia. The incidence of both health issues has been observed to be strongly interlinked where presence of one exacerbates the other. Nonalcoholic fatty liver disease (NAFLD) pathophysiology increases the muscle loss, while the onset of NAFLD in sarcopenic patients aggravates the liver problems as compared to non-sarcopenic patients. Scarcity of research on the subject provides very little evidence about the cause and effect of disorders. No FDA approved drugs are available to date for NAFLD and sarcopenia. Research is underway to understand the complex biochemical pathways involved in the development of both disorders. This review is a small contribution toward understanding sarcopenia in the setting of NAFLD that provides insight on the common pathophysiological profile of sarcopenia and NAFLD and portrays a novel way of delving into the subject by introducing the concept of cortisol crosstalk with the muscle-liver axis. Sarcopenia and NAFLD are considered metabolism-related problems, and cortisol, being a glucocorticoid, plays an important role in metabolism of fats, carbohydrates, and proteins. Cushing’s syndrome, characterized by abnormally elevated concentrations of blood cortisol/enhanced intracellular activity, shares many pathologic conditions (such as insulin resistance, metabolic syndrome, abnormal levels of specific cytokines, and obesity) with NAFLD and sarcopenia. Hence, cortisol can be a potential biomarker in sarcopenia and NAFLD. As cortisol activity at cellular level is controlled by 11β-hydroxysteroid dehydrogenase type 1 and 2 (11β-HSD1/2) enzymes that convert inactive steroid precursor into active cortisol, these enzymes can be targeted for the study of sarcopenia and NAFLD. Combined studies on NAFLD and sarcopenia with respect to cortisol open a new avenue of research in the understanding of both disorders.
Keywords
INTRODUCTION
The term “sarcopenia” was introduced by Irwin Rosenberg to refer to the loss of muscle mass in older people[1]. For a long time, sarcopenia was considered as an age-related disorder that affected the quality of life among the elderly population[2]. Sarcopenia is known currently as a progressive skeletal muscle disorder of multifactorial origin. It involves the waste of muscle mass and function, linked to a higher risk of adverse outcomes, such as fractures, physical disability, falls, and fatalities[3]. The categories of primary and secondary sarcopenia have been employed in clinical practice. Primary sarcopenia is largely attributed to aging, while secondary is due to other causes, such as type 2 diabetes mellitus (T2DM), osteoporosis, cancer, cardiovascular disease, metabolic syndrome (MetS), and obesity[4].
A substantial number of meta-analyses and both longitudinal and cross-sectional studies in healthy and diseased individuals have demonstrated a strong association of sarcopenia with nonalcoholic fatty liver disease (NAFLD)[5-9]. NAFLD includes a broad spectrum of liver disorders, ranging from triglycerides (TGs) accumulation in hepatocytes or simple steatosis, progressing to inflammatory hepatocellular and ballooning injury, a condition known as nonalcoholic steatohepatitis (NASH). Further progress eventually leads to cirrhosis and advanced fibrosis, conferring a higher risk of liver cancer[10-12].
Sarcopenia and NAFLD are considered obesity-related disorders[13]. There is compelling evidence that skeletal muscle, liver, and adipose tissue behave as glands, showing endocrine, autocrine, and paracrine activities[13-15]. Secretion of cytokines and other signaling molecules constitute the basis of the molecular crosstalk along the muscle-liver-adipose tissue axis, where cortisol may pose an important role as a key modulator.
This review is concerned with the recent progress that has been made in understanding the association of sarcopenia with NAFLD and the involvement of cortisol in the pathophysiological mechanisms underlying these disorders.
SARCOPENIA
Skeletal muscle tissue has a central role in human locomotion, and, as an endocrine organ, it aids in maintaining metabolic homeostasis[16]. It represents almost half of the human body mass, and any malfunctioning in its properties has a considerable impact on health. Practically 45% of the human body mass comprises muscle mass and people lose an estimated 1%-2% of muscle mass per year after 50 years of age[17].
Assessment of muscle strength, muscle mass, and performance is necessary for clinical trials to diagnose sarcopenia. Many studies have investigated the prevalence of sarcopenia in the world. However, due to the variety of assessment techniques and cutoff points, the results are inconsistent[3,18]. A systematic review and meta-analysis study estimated the overall prevalence of sarcopenia in both genders from different world regions[19]. The results reveal that a substantial proportion of older people have sarcopenia, even in healthy populations, and that the overall prevalence was the same for both sexes (10%). However, variations in prevalence were found among non-Asian and Asian individuals, being higher among non-Asians. Additionally, significant variations are associated with different populations (community-dwelling, hospitalization, and living in nursing homes), making an early diagnosis that can prevent adverse outcomes more difficult[20].
Etiology
The etiology of sarcopenia is not fully understood. Sarcopenia is predominantly a geriatric condition, but young and middle-aged people are at risk of developing it due to multiple factors that are not primarily related to aging per se. Malnutrition, diabetes, hormonal imbalances, low physical activity, or comorbidities such as cancer[21], chronic respiratory diseases[22], mental diseases[23], hepatic diseases[24], renal failure[25], Cushing’s syndrome[26], genetic predisposition, and the effects of environment in early development are all associated with the development of muscle wasting[27,28].
Various modifications during aging play a fundamental role in the pathogenesis of primary sarcopenia. Hormonal changes, chronic inflammation, mitochondrial abnormalities, loss of neuromuscular junctions, metabolic disturbances, and nutritional deficiencies can trigger the loss of muscle mass, strength, and functionality typical of sarcopenia. In elderly persons, there is a significant association among sarcopenia, increased levels of proinflammatory cytokines, and the reduction in relevant anabolic hormones. Chronic low-grade inflammation is a consequence of the plasma increase of proinflammatory mediators, such as tumor necrosis factor α (TNF-α), interleukin-6 (IL-6), and C-reactive protein (CRP)[29]. Other circulating factors, such as TNF-like weak inducer of apoptosis, IL-18, leptin, insulin growth factor 1 (IGF1), insulin, and adiponectin appear associated with sarcopenia[30,31].
Age-related changes in redox homeostasis also seem to play a fundamental role in sarcopenia. Oxidative stress and the accumulation of reactive oxygen species (ROS) increase with aging, causing severe damage to muscle cells[32]. An imbalance in the generation of ROS and the subsequent oxidative damage is associated with mitochondrial dysfunction in skeletal muscle aging[33]. The decrease in mitochondrial function results in bioenergetic insufficiency[34], increased apoptosis, and reduced capacity for muscle regeneration, finally leading to muscular atrophy and muscle denervation[35,36].
Metabolic disorders are important factors affecting the age-related loss of skeletal muscle[37]. Insulin resistance (IR), diabetes, MetS, and obesity are associated directly with primary sarcopenia and sarcopenic obesity. Sarcopenic obesity (SO) refers to the association and co-occurrence of obesity and sarcopenia, synergistically reinforcing each other to increase muscle loss, fat mass, and IR[38,39]. Insulin plays a major role in regulating muscle protein metabolism and mitochondrial oxidative phosphorylation in human skeletal muscle. IR is involved in the age-related muscle protein loss that progressively leads to sarcopenia[40,41].
Treatment
Currently, there is no US Food and Drugs Administration (FDA)- or European Medicines Agency (EMA)-approved medication for the treatment of sarcopenia. However, several therapeutic approaches have been explored, including hormone replacement, selective modulators of the androgen receptor (SARMs), myostatin, and angiotensin-converting enzyme (ACE) inhibitors.
Hormone replacement has received considerable interest as a therapy for sarcopenia. However, inconsistent results and a high risk of severe secondary effects have contributed to dismissing hormone replacement therapy from clinical use. Some trials with estrogen, testosterone, growth hormone, and growth hormone-releasing hormone therapies showed significant increases in lean body mass and muscle strength, while others failed to yield any meaningful increase in muscle strength[42].
SARMs are employed as a potential alternative to testosterone therapy for treating muscle wasting associated with age-related sarcopenia, as well as several other conditions, including end-stage liver disease, chronic infection, immobilization, and chronic glucocorticoids (GCs) use[43]. SARMs are chemically engineered small molecule drugs that can selectively exert varying degrees of agonist and antagonist effects on the androgen receptor throughout the body without androgenic side effects. Clinical trials showed that various SARMs (GSK2881078 and enobosarm) are effective in ameliorating muscle wasting by increasing the lean body mass[44-46], although no improvement in physical function has been recorded[47].
Another therapeutic approach for treating sarcopenia includes the use of myostatin inhibitors. Myostatin is a member of the transforming growth factor-β (TGF-β) family. It binds to the type IIB activin receptor (ActRIIB), activating the signaling cascade that leads to inhibition of myoblast differentiation and proliferation[48].
Overexpression of the myostatin gene leads to reduced muscle mass, suggesting it is a negative regulator of skeletal muscle growth. Additionally, myostatin regulates bone metabolism by suppressing bone formation and stimulating bone resorption[49]. Myostatin is also associated with fat accumulation and adipogenesis[50]. In addition to an increased lean mass, myostatin-deficient mice show a diminished fat mass and increased insulin sensitivity and glucose uptake[51], apparently as an indirect consequence of changes in skeletal muscle metabolism[52]. In humans, high myostatin levels are positively correlated with body weight, body mass index, and waist and hip circumference[53-55] and decrease after aerobic exercise training[56].
Several myostatin inhibitors have been developed for various musculoskeletal disorders, and many of them are currently under evaluation in human trials[49,57]. Clinical trials have demonstrated that inhibition of myostatin/ActRIIB signaling by myostatin antibodies, anti-myostatin peptibodies, activin A antibodies, soluble forms of ActRIIB recombinant fusion proteins, anti-myostatin adnectin, and ActRIIB antibodies may help to improve muscle mass in patients with muscle wasting[57]. The principal drawback of developing myostatin inhibitors is that most of them also block the activity of other closely related members of the TGF-β family due to their high sequence and structural similarities.
Some of the compounds developed as myostatin inhibitors have been discontinued due to the absence of significant improvements in muscle function. Others look promising, such as bimagrumab, a fully human monoclonal antibody that binds to ActRII. Recent studies showed that bimagrumab leads to a marked loss in fat mass, an increase in lean mass, and improvement in a range of metabolic biomarkers, with mild or moderate adverse events that did not require treatment and resolved spontaneously during the follow-up period[58,59].
ACE inhibitors and angiotensin receptor blockers (ARBs) have been proposed as a treatment for improving peripheral skeletal muscle function. Sarcopenia is highly prevalent in hypertensive older adults, and ACE inhibitors seem to have favorable effects on both disorders[60,61]. However, results from two systematic reviews and meta-analysis reveal that there is not enough evidence for using ACE inhibitors or ARBs as stand-alone therapies to improve physical performance in older people, either with or without a
At present, the therapeutic options for the management of sarcopenia are physical exercise and nutritional interventions (dietary supplements or nutraceuticals)[64,65]. Resistance training improves the power-producing capacity of skeletal muscle fibers, increases insulin sensitivity, and sensitizes muscles to other anabolic stimuli counteracting the anabolic resistance. Endurance exercise increases the oxidative capacity of skeletal muscle by a rise in mitochondrial biogenesis and supports faster protein turnover during resistance training[66]. Additionally, aerobic exercise and resistance training downregulate circulating inflammatory biomarkers, including IL-6, CRP, and TNF-α[67,68]. Consequently, a combination of endurance and resistance exercises is an effective measure for enhancing the quality of life in the elderly by improving skeletal muscle functional capacity and plasticity[69].
Current evidence suggests that the intake of high-quality protein or nutritional supplements enhances the increased muscle mass, strength, and function induced by resistance training. Nutritional supplementation includes free leucine (the most potent amino acid for the stimulation of muscle protein synthesis), creatine, vitamin D, and omega-3 fatty acids[70,71]. Older adults may require higher amounts of protein than the current recommendations to counteract anabolic resistance. A systematic review and meta-analysis of observational studies indicate that a very high (≥ 1.2 g/kg/day) or high (≥ 1.0 g/kg/day) protein intake compared to a low protein intake (< 0.8 g/kg/day) is associated with a better lower-limb physical performance in community-dwelling older adults[72].
The efficacy of nutritional supplements by themselves in ameliorating sarcopenia is less clear. Nutraceuticals and pharmaceutical interventions are neither as effective nor show synergistic effects when administered concurrently with exercise regimes[36,73].
Different approaches for developing new treatments are currently underway. One of them concerns the development of compounds that produce the effects of physical exercise (exercise mimetics or exercise pills) for subjects who cannot tolerate or are unwilling to undertake physical activity. The use of natural products has been an object of study in experimental animals and humans. Recent studies in animal models suggest that supplementation with natural compounds, such as ursolic acid (enriched in apples), tomatidine (abundant in unripe tomatoes), and urolithin A and ellagitannins (found in pomegranates, as well as nuts and berries), may ameliorate sarcopenia in older individuals[74]. A systematic review of studies from 2010 to 2015 that correlated botanical extracts to skeletal muscle health suggests that curcumin from Curcuma longa, alkaloids and steroidal lactones from Withania somnifera (Solanaceae), catechins from Camellia sinensis, proanthocyanidin of grape seeds, and gingerols and shogaols from Zingiber officinale may be potentially useful dietary supplements to prevent loss of muscle mass and function[75].
Promising therapeutic strategies for future management of sarcopenia involve CRISPR techniques[76], modulation of inflammatory signal pathways through the use of microRNA[77], and stem cell therapy[36,78,79].
NAFLD
With a worldwide prevalence over the last two decades of nearly 25% of the adult population, NAFLD has emerged as the most common liver disorder and a leading cause of cirrhosis and hepatocellular carcinoma[80]. As currently accepted, NAFLD represents the liver contribution to a cluster of conditions associated with metabolic dysfunction[81]. There is also evidence that NAFLD is the hepatic manifestation of MetS. NAFLD coexists with impaired glucose tolerance, central obesity, dyslipidemia, and hypertension, which represent the main features of MetS[82]. NAFLD encompasses a broad spectrum of disease severity and inter-patient variability in disease progression and response to treatment. Multiple factors, including age, gender, ethnicity, diet, genetic predisposition, and metabolic status, among others, are likely to influence this heterogeneity in both the clinical presentation and the course of the disease[83].
NAFLD is characterized by the presence of steatosis in more than 5% of hepatocytes in association with metabolic risk factors, notably obesity and T2DM, and in the absence of other chronic liver diseases[84,85]. Steatosis can occur with and without mild inflammation [nonalcoholic fatty liver (NAFL)]. In a fatty liver, micro- and macrovesicles [lipid droplets (LDs)] of TGs assemble within the hepatocytes without causing considerable inflammation, scarring, or cell death[86]. The disease may progress through the NASH necroinflammatory condition, which is also characterized by hepatocellular damage associated with hepatocyte ballooning. As the disease progresses, the accumulating liver fibrosis will eventually lead to cirrhosis, hepatocellular carcinoma, and liver failure[11].
The remission of NAFL can be relatively easily achieved[87]. The disease is stable or progresses slowly in most NAFLD patients, and it will not end in cirrhosis or liver-associated death. However, a small proportion of patients develop advanced fibrosis that may progress to end-stage liver disease and hepatocellular carcinoma[81]. The development of fibrosis occurs at a much higher rate in NASH patients, presumably driven by necroinflammation[88]. Fewer than 10% of patients with NAFLD develop cirrhotic complications and hepatocellular carcinoma during the 10-20 years after diagnosis. However, given the high disease prevalence, the mortality is substantial in absolute numbers[89,90]. Cirrhosis is the highest risk factor for developing hepatocarcinoma, with an annual incidence rate of about 11 per 1000 person-years[91]. Despite the progressive risk of liver disease, the leading cause of fatality in NAFLD patients is cardiovascular disease (CVD), followed by extrahepatic malignancies such as colorectal or breast cancer. These causes of death are likely due to common cardiometabolic risk factors in NAFLD and CVD, although it is not yet clear to what extent NAFLD contributes directly to the development of CVD[81].
Etiology
The main driver of NAFLD is overnutrition that leads to expansion of adipose depots and accumulation of ectopic fat. The infiltration of macrophages in visceral adipose tissue results in a proinflammatory state that promotes IR. Under these conditions, inappropriate lipolysis leads to sustained delivery of fatty acids (FAs) to the liver, which, in addition to an increased de novo synthesis of lipids, exceeds its metabolic capacity[81]. The imbalanced lipid metabolism leads to formation of lipotoxic compounds that contribute to cellular stress, inflammasome activation, and apoptosis, with the subsequent stimulation of inflammation, tissue regeneration, and fibrogenesis[81,92-94].
Multiple metabolic, genetic, epigenetic, and microbiome-associated factors influence the disease[13,94]. The heritable NAFLD component shows inter-individual variations[95]. Five common genetic variants are associated with NAFLD[96]. A single-nucleotide polymorphism (cytosine to guanine substitution) in the PNPLA3 gene (PNPLA3 I148M variant) is the best-characterized genetic variant associated with susceptibility to NAFLD and is strongly related to increased hepatic fat levels, hepatic inflammation, and raised liver-disease mortality[94,97,98].
Treatment
An altered body energy balance is critical for the development of NAFLD and NASH. The improvement in diet quality and sustained or increased physical activity reduces the risk of developing NAFLD, even among high genetic risk individuals[99,100]. Existent data indicate that the loss of body weight reduces the NAFLD progression. The disease can revert readily in patients with mild NAFLD, and a considerable improvement in NAFLD severity is achieved after lifestyle modification and weight loss. The hepatic fat content of NASH patients is reduced more than 5% by body weight loss, and fibrosis decreases when this loss exceeds 10%, irrespective of the method employed[101]. However, the use of drugs to reduce IR and hyperlipidemia is highly recommended in more advanced NAFLD stages[102].
The FDA and EMA have no drugs approved yet for direct NAFLD or NASH therapy. Various drugs currently available for other disorders have been under clinical trials for NAFLD and NASH with limited active participants (50-100), targeting mainly energy metabolism. These include pioglitazone (a thiazolidinedione) that might prevent the development of T2DM[103], vitamin E, and two classes of glucose-lowering agents for the treatment of T2DM: glucagon-like peptide-1 (GLP-1) receptor agonists (liraglutide and semaglutide) and sodium-glucose cotransporter type 2 (SGLT2) inhibitors (canagliflozin, dapagliflozin, and empagliflozin). They all improve hepatic steatosis and necroinflammation. Pioglitazone and vitamin E might induce serious adverse effects such as the increased risk of bladder and prostate cancer, bleeding risk, heart failure, and hemorrhagic stroke at high vitamin E doses[81]. However, GLP-1 receptor agonists and SGLT2 inhibitors show promising results, particularly semaglutide that exhibits a notably high NASH resolution rate[81]. Numerous drugs with different mechanisms of action, targeting energy intake, energy disposal, lipotoxic hepatic injury, and the resulting inflammation and fibrogenesis, are being developed as a treatment for NASH[104].
Disease drivers can vary notably with NAFLD patients, and both the disease progression and the response to treatment are heterogeneous[81,83]. Although NAFLD is not a life-threatening condition, its high prevalence rate and association with other severe morbidities such as T2DM, hyperlipidemia, hypertension, IR, MetS, and obesity is a cause of concern.
CORRELATION OF SARCOPENIA AND NAFLD
Sarcopenia has been regarded as a typical aging problem, although many studies provide evidence of its relationship with several common metabolic disorders. The association between muscle dysfunction and NAFLD, two obesity-related disorders, is primarily based on a sustained caloric excess that disrupts the muscle-liver-adipose axis mechanism leading to end-stage tissue damage typified by cirrhosis and sarcopenia[13].
Many studies with cohorts of patients from different world regions have focused on the bidirectional relation between sarcopenia and NAFLD. Early reports have shown the impact of a low muscle mass on NAFLD. A prospective observational study with 452 participants demonstrated an increased risk of NAFLD in adult patients with low muscle mass[5]. A reduction in muscle mass is associated with the severity of liver histological changes, as observed in a study with 309 NAFLD patients[6]. There is an inverse relationship between skeletal muscle mass index (SMI) and incident NAFLD in about 15% of the whole population of a longitudinal study over seven years with 15,567 subjects[8]. A positive relationship between an increased SMI with NAFLD resolution is also observed in 2.7% of 2943 NAFLD subjects, suggesting a favorable impact of muscle mass on NAFLD[8].
NAFLD progression increases among patients with low SMI and low muscle strength, as shown recently in a prospective study with 5132 participants aged 18-80 years. The NAFLD progression becomes stronger with sarcopenia and more so with SO, demonstrating that the coexistence of sarcopenia and obesity leads to a higher risk of NAFLD[105]. Cai et al.[7] also reported an inverse relation of SMI with NAFLD in a recent meta-analysis, lending further support to the existence of a low muscle mass-NAFLD direct relationship. Interestingly, SMI appears negatively correlated with hepatic steatosis exclusively among male but not female T2DM patients[106], stressing the importance of a differential hormonal effect on the
Sarcopenia is associated with a two-fold risk of developing significant fibrosis, independent of obesity and IR[107]. Significant liver fibrosis is more prevalent in sarcopenic subjects than in those without sarcopenia (45.7% vs. 24.7%)[6]. Likewise, cross-sectional data analysis of 11,325 American participants of the third NHANES survey shows that NAFLD is more prevalent in individuals with sarcopenia than in those without (46.7% vs. 27.5%). Moreover, advanced liver fibrosis is more common in patients with sarcopenia than in those without (7.8 vs. 1.6%). These data indicate that sarcopenia in NAFLD represents a risk factor of advanced fibrosis independent of metabolic risk factors[108]. Cai et al.[7] also reported a strong association of sarcopenia with advanced stages of NAFLD and NASH. Additionally, they showed that the recovery of skeletal muscle mass might help in preventing the development and progression of NAFLD in healthy as well as in sarcopenic patients[7].
Conversely, some studies have shown that NAFLD influences the onset of sarcopenia. A cross-sectional analysis of the Korean National Survey (2008-2011) of 2761 adults of both genders with NAFLD reports a 12.2% sarcopenia incidence, diagnosed by dual-energy X-ray absorptiometry, the gold standard for measuring body fat[107]. A wealth of data shows the detrimental effects of advanced NAFLD on sarcopenia. The prevalence of sarcopenia rises as the liver disease progresses from 8.7% to 17.9% and 35.0% in non-NAFLD, steatosis, and NASH, respectively. Sarcopenia appears significantly associated with NASH and significant fibrosis, independent of obesity, inflammation, and IR, confirming the above findings of
A study from Canada analyzed the presence of sarcopenia, SO, and myosteatosis (muscle fatty infiltration) in a cohort of 678 cirrhotic patients of different etiologies, including chronic hepatitis C (40%), alcohol (23%), and NASH/cryptogenic (14%)[109]. Myosteatosis was reported in 52% of patients, while sarcopenia and SO were found in 43% and 12% of subjects, respectively. The median survival time of patients with myosteatosis, sarcopenia, and SO (about 23 months) was considerably shorter than that of patients having no muscular abnormalities (92 months). Multivariate analysis evidenced a raised mortality risk associated with myosteatosis and sarcopenia in cirrhotic patients (HR = 1.42 and 2.00, respectively)[109]. In cirrhotic patients undergoing liver transplant, myosteatosis has been related to more clinical complications and poor survival[13]. Sarcopenia helps predicting adverse clinical outcomes in patients with NAFLD suffering from cirrhosis, such as liver decompensation, poor quality of life, and early fatality[110].
The above studies evidence the existence of a bidirectional relationship between loss of muscle mass and NAFLD progression, while the presence of obesity, especially visceral obesity, is an important key player, as studies indicate that the disruption of the muscle-liver-adipose axis mechanism plays a significant role in the progression of both sarcopenia and NAFLD. A population-based longitudinal study for 10 years with 8399 participants in Japan reported the visceral obesity index as a predictor of NAFLD[111]. The presence of adipokines plays an important role in aggravating the chronic inflammation observed in NAFLD. Intra-abdominal fats are worth mentioning as an origin of these adipokines[112]. In a study comprised of 142 NAFLD patients, liver biopsies were conducted, and visceral adiposity index was used as a clinical measurement with the assessment of serum levels of different cytokines. The study found the independent correlation of insulin resistance and visceral adiposity index with significant fibrosis in NAFLD[113]. A review of 24 studies reported the visceral adiposity index as a predicting factor in the diagnosis of NASH and NAFLD and a higher value of visceral adiposity index in NAFLD patients as compared to persons without NAFLD[114]. A study with 352 participants found a correlation between NAFLD onset and thickness of visceral fats[115]. Francque et al.[116] reported a considerable association of visceral obesity with the degree of steatosis.
In a retrospective study of 250 lean NAFLD patients, the association of visceral obesity and hepatic steatosis was investigated. It found a positive correlation between NAFLD and visceral obesity in lean Asian population and visceral adiposity was determined as a risk factor for NAFLD severity[117]. Some Chinese researchers analyzed the relationship between the incidence of NAFLD and visceral obesity. The baseline data of 4809 subjects and results of four follow-up examinations were included in the cohort study. The cumulative incidence of four-year follow-up was 13.9%[118]. A Japan-based cross-sectional study with 3197 participants investigated the association of NAFLD with visceral and subcutaneous adiposity. It was reported that NAFLD plays an important role in the development of obesity[119]. Researchers from Turkey examined the relationship of clinical and histological parameters in NAFLD cases with the visceral obesity index. A control group of 57 patients was compared with the experimental group. Significantly higher values of VAI were observed in the experimental group as compared to the control group and values were even more noticeable in the case of NASH[120].
NAFLD PATHOPHYSIOLOGY, SARCOPENIA AND MYOSTEATOSIS
Another parallel in the progression and severity of NAFLD is myosteatosis along with sarcopenia according to many studies. A longitudinal study was conducted on 184 patients with morbid obesity to check the correlation of myosteatosis with NAFLD. Researchers concluded that skeletal muscles fat content is strongly linked with the severity of disease, especially NASH. Fat content in muscles decreases upon improvement in NASH. Therefore, fat infiltration of skeletal muscles is an important marker in NAFLD[121]. Although knowledge about the clinical significance of muscles fat infiltration in NAFLD is scarce, many common underlying mechanisms provide a strong basis for their association. A common factor is insulin resistance that crosstalks with NAFLD, sarcopenia, and myosteatosis[122]. Studies report that myosteatosis is more common in NAFLD as compared to other liver problems. Moreover, adverse muscle composition (myosteatosis and low muscle volume) is very frequent in NAFLD patients[4]. According to a study in Expert Review of Gastroenterology & Hepatology, sarcopenia was associated with adverse events such as poor life quality, premature mortality, and hepatic decompensation[110]. A study on rodent models assessed the effect of low muscle, myosteatosis, and low muscle strength on NAFLD severity and NASH. The study reported a substantial link of myosteatosis with inflammation and hepatocellular damage during the early development of NASH that makes myosteatosis a novel marker of NASH[123].
A cross-sectional study of 9545 participants was conducted in Sweden to determine the association of adverse muscle composition with NAFLD. Different factors such as free muscle volume, liver fat, and muscle fat infiltration were determined by MRI. Out of all participants, 1204 had NAFLD issues, and, among NAFLD patients, 14% had adverse muscle composition[124]. Myosteatosis is not only linked to NAFLD problems but has a profound effect on other liver problems as well. A follow-up study of 48 obese patients showed a strong correlation of liver stiffness with myosteatosis and a decrease in myosteatosis after dietary intervention improved liver stiffness[125]. In another retrospective cross-sectional study, researchers investigated the possible link of myosteatosis and sarcopenia with different stages of NAFLD. The study included 48 obese patients. The incidence of sarcopenia was only 8.3%, while skeletal muscle fat index was relatively higher in patients with liver stiffness. These results provide a strong basis of myosteatosis association with liver stiffness in NAFLD and a sharp reduction in liver stiffness with a decrease in myosteatosis after three months[126]. Another cross-sectional study with 5132 participants focused on investigating the correlation of muscle-related issues with NAFLD. Low muscle strength and low muscle mass were common in NAFLD patients. The incidence of NAFLD was higher in patients with sarcopenia, while the presence of both obesity and sarcopenia put the patients at higher risk of severe NAFLD[105].
Sarcopenia and myosteatosis are also linked with liver problems other than NAFLD that shows their key role in liver problems. A Canadian review study reported a high prevalence of myosteatosis and sarcopenia in patients with liver abnormalities such as cirrhosis[127]. In a Korean cohort, it was found that the severity of fibrosis in NAFLD is strongly associated with visceral fat content and quality and quantity of muscles. A follow-up study of 521 NAFLD assessed the skeletal muscles index, visceral adipose tissue index, and muscle attenuation via computed tomography. Out of all cirrhosis patients, 45.1% had low skeletal muscle mass, 46.1% had myosteatosis, and 46.9% had visceral adiposity. Hence, myosteatosis, low skeletal muscle mass, and visceral adiposity are independent risk factors for fibrosis in NAFLD patients[128].
NAFLD PATHOPHYSIOLOGY AND MUSCLE LOSS
The overlap in the pathophysiology of NAFLD and sarcopenia is widely recognized. However, the specific pathways implicated in the pathogenesis of these diseases are still unclear. It is unknown whether sarcopenia is the cause or consequence of NAFLD progression. IR, obesity, physical inactivity, aging, and specific cytokines are at the base of the pathophysiology of both disorders[129] [Figure 1].
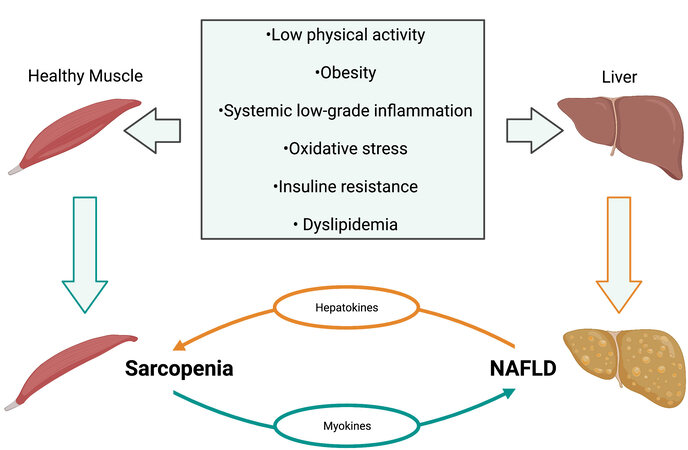
Figure 1. Common risk factors between sarcopenia and NAFLD. The figure depicts the most common etiologies of both diseases and their reinforcing relation. NAFLD: Nonalcoholic fatty liver disease.
Liver, skeletal muscle, and adipose tissue are essential organs involved in metabolic energy storage and regulation of systemic energy homeostasis that can sense and respond to nutrient demands through multiple crosstalks between themselves and other tissues. The control of systemic energy homeostasis is achieved in part by the regulation of glucose and lipid metabolism. Dysregulation of either process can lead to metabolic dysfunction and contribute to the development of IR[130].
The liver is the main organ for the storage of metabolic energy as glycogen and TGs and is essential in the control of systemic glucose homeostasis. Additionally, it has the unique property of exporting glucose in response to energy demands[131]. The true character of NAFLD refers to the accumulation of TGs in the liver, which are the esterification products of glycerol and free FAs. These FAs arise from different sources that include diet, lipolysis, and de novo lipogenesis. The body either utilizes FAs through re-esterification to TGs, β-oxidation, or storage in the form of LDs or exports them as very-low-density lipoproteins. Any imbalance in these processes raises the liver content of TGs. Therefore, decreased fat export, increased fat uptake or synthesis, or decreased fat oxidation can result in fat accumulation within the hepatocytes, leading to NAFLD[132].
Under hyperinsulinemic conditions, the free FAs efflux from the expanding adipose tissue mass leads to an enhanced fat uptake by the liver[133]. In addition, insulin continues to activate lipogenesis, enhancing FAs synthesis and hepatic TGs accumulation, further driving NAFLD[134]. Hepatic FAs uptake and de novo lipid synthesis increase even more as the oxidation and export of FAs cannot compensate for such a rise. This situation may result in cellular damage and disease progression by inducing oxidative stress with compromised mitochondrial and peroxisomal oxidative activity[135].
During chronic hyperinsulinemia, there is an increase in adipose tissue inflammation and in local and hepatic IR that occurs via the production of proinflammatory cytokines, mainly TNF-α and IL-6, which in turn causes ectopic fat deposition in many organs. Insulin-resistant adipose tissue thus creates a vicious cycle of accumulating lipotoxic metabolites, steatosis, and IR[136,137]. Hyperinsulinemia, hepatic IR, and steatosis are caused by augmented JNK-1 signaling in the adipose tissue[136,137]. JNK-1 is an essential element of a metabolic stress signaling pathway that regulates IL-6 expression in fatty tissue[138]. NAFLD and NASH patients have serum IL-6 levels significantly greater than healthy individuals[139]. Due to the pleiotropic nature of IL-6 in the regulation of inflammation and metabolism, a chronic low-grade inflammation mediated by IL-6 in adipose tissue can be related to the progression of NAFLD[140,141].
Skeletal muscle also utilizes lipids, in addition to glucose, as important energy substrates. However, increased lipid overload and free FAs influx might impair insulin signaling[142]. High levels of lipid metabolites such as long-chain fatty acyl-CoAs, diacylglycerols, and ceramides can activate intracellular kinases that promote inhibitory serine/threonine phosphorylation of the insulin receptor and insulin receptor substrate 1 (IRS1), affecting the insulin action in skeletal muscles[143]. Skeletal muscle also express pro-inflammatory molecules in response to lipid excess, contributing to low-grade systemic inflammation and IR[144]. Early studies have shown that TNF-α can induce inhibitory phosphorylation of the IRS1 and decrease the stimulatory phosphorylation of Akt kinase, promoting IR and compromising glucose metabolism in skeletal muscles[145]. Muscle metabolism is impaired and proteolysis is triggered, resulting in a progressive loss of muscle mass.
Dysregulation of glucose and lipid metabolism responds to the multiple crosstalks among liver, skeletal muscle, and adipose tissue and with other tissues through different factors and cytokines, contributing to the development of IR [Figure 2].
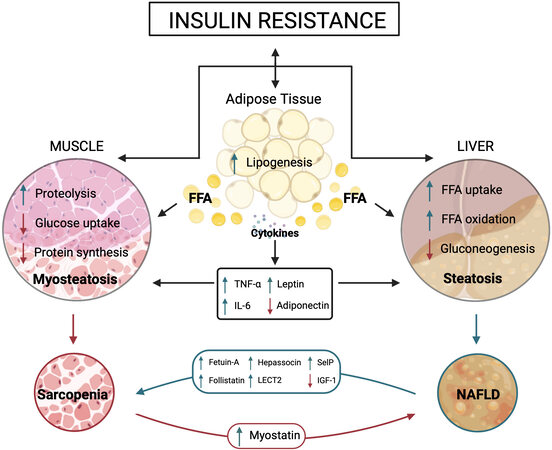
Figure 2. Crosstalk among adipose tissue, liver, and skeletal muscle. The figure shows the main adipokines, hepatokines, and myokines interplaying with target organs under insulin resistance condition. ↓In red represents a decrease and ↑in green represents an increase in action. FFA: Free fatty acids; TNF-α: tumor necrosis factor alpha; IL-6: interleukin-6; LECT2: leukocyte cell-derived chemotaxin 2; SelP: selenoprotein P; IGF-1: insulin growth factor; 11β-HSD1: 11β-hydroxysteroid dehydrogenase type 1; 11β-HSD2: 11β-hydroxysteroid dehydrogenase type 2.
IR, in turn, affects the expression of some adipokines. Thus, adiponectin secretion decreases while that of leptin increases. High-leptin serum levels promote hepatic inflammation and fibrogenesis and have been linked to NAFLD progression in experimental and clinical studies[146,147]. In contrast, serum adiponectin levels become reduced in patients with NAFLD, NASH, obesity, and T2DM[148-150]. Leptin directly affects glucose and FAs metabolism in skeletal muscle through the insulin signaling pathways at the levels of IRS and phosphatidylinositol-3 kinase[151]. In addition, leptin directly activates 5′-AMP-activated protein kinase (AMPK) in skeletal muscle, holding anabolic pathways and enhancing catabolism in response to a decrease in the ATP/AMP ratio[152,153].
The liver secretes an ample number of hepatokines. Several of them show a positive correlation with IR, are increased in NAFLD, and are directly implicated in the liver-muscle crosstalk[130,154]. This group comprises angiopoietin-like proteins, fetuin-A, follistatin, hepassocin, leukocyte cell-derived chemotaxin 2 (LECT2), and selenoprotein P [Figure 2].
Fetuin A is an inhibitor of the insulin receptor tyrosine kinase in the liver and skeletal muscle. The plasma fetuin-A levels correlate positively with high liver fat content, early atherosclerosis, and MetS in middle-aged and elderly patients[155,156]. In addition, fetuin-A induces IR and inflammation via multiple mechanisms, including inhibition of insulin-sensitizing adiponectin production[157].
Follistatin (FST) is a multi-faceted glycoprotein that is involved in multiple functions. Although secreted by a broad range of cell types, the liver contributes mainly to the circulating FST levels in humans[158]. FST is acutely regulated by the glucagon-to insulin ratio. Thus, glucagon is stimulatory, whereas insulin inhibits FST secretion in vivo and in vitro, mediated via cAMP in the hepatocyte[158]. It inhibits members of the
Hepassocin (HPS) is another hepatokine involved in skeletal muscle IR through an AMPK-dependent mechanism. Increased expression of HPS in hepatocytes contributes to the development of IR in skeletal muscle via the EGFR/JNK-mediated pathway[161].
LECT2 is implicated in NAFLD, and its expression is positively correlated with body weight and IR in humans. LECT2 activates JNK in myocytes and inhibits insulin signaling via increased serine phosphorylation of IRS1[162].
Finally, selenoprotein P (SelP) is a hepatokine usually involved in selenium transport. However, its secretion increases in NAFLD, prediabetes, and T2DM. It contributes to hyperglycemia in T2DM by inducing IR in the liver and skeletal muscle. SelP impairs insulin signaling and dysregulates glucose metabolism in hepatocytes and myocytes via AMPK inhibition[163].
In contrast to the effects of preceding hepatokines, IGF1 levels decrease in patients with obesity and NAFLD. Low plasma levels of IGF1 are correlated with IR and increased risk of MetS[164]. IGF1 is secreted into the circulation by the liver in response to growth hormone stimulation (GH)[165]. IGF1 acts primarily on skeletal muscle, where it elicits its insulin-sensitizing effects[166]. The binding of IGF1 to its receptor (IGF1R) phosphorylates intracellular adaptor proteins and activates MAPK and PI3K/AKT signaling pathways. IGF1 is regulated by an IGF-binding protein (IGFBP) with equal or greater affinity for IGF1 than that of IGF1R. IGF1BP1 levels are increased in T2DM patients and counteract the hypoglycemic effect of insulin through IGF1 inhibition.
IGF1 administration improves insulin sensitivity and lowers glucose levels in patients with and without T2DM. In contrast, a reduction in IGF levels occurs in patients with obesity and NAFLD, consistent with an inhibitory effect of high levels of free FAs on the GH/IGF-1 axis, which generally plays a protective role in age-related muscle loss and muscle regeneration[167-169].
Skeletal muscle secretes myokines involved in the pathophysiological mechanisms of NAFLD. Myonectin, irisin, and IL-6 increase in response to physical exercise and protect against NAFLD development by preventing obesity-induced lipid accumulation and associated IR. Aerobic exercise suppresses the expression of myostatin[56]. Initially discovered as a regulator of skeletal muscle mass, myostatin has hepatic effects due to its action on skeletal muscle metabolism. Decreasing myostatin levels not only increase muscle mass but also protect from fatty liver and improve IR[51,170]. Myostatin levels are significantly elevated in patients with end-stage liver disease and associated with a reduced overall survival rate in liver cirrhosis patients[171,172].
ROLE OF CORTISOL SIGNALING IN SARCOPENIA AND NAFLD
Cortisol, a member of the GCs family, is a steroid hormone secreted by the adrenal cortex that plays different roles. Its secretion follows a circadian rhythm destined to control tissue repair, immune stability, and metabolic processes. In response to stress conditions, there is an elevated and transient release of cortisol that allows regulation of lipid, carbohydrate, and protein metabolism for maintaining adequate glucose levels in the brain and other organs.
GCs secretion is under the control of the hypothalamic-pituitary-adrenal (HPA) axis. The hypothalamus secretes corticotropin-releasing hormone (CRH) in response to physiological or psychological stress. CRH stimulates the anterior pituitary gland to secrete the adrenocorticotropic hormone (ACTH), which induces the synthesis and secretion of cortisol. CRH and ACTH synthesis and release are inhibited by cortisol-mediated negative feedback, contributing to the suppression of the HPA axis regulation of GCs levels. The concentration of active GCs can be modulated locally by two 11-β-hydroxysteroid-dehydrogenases
During stress conditions, cortisol affects the metabolism to elevate blood glucose by mobilizing metabolites through gluconeogenesis and glycogenolysis. Lipolysis from peripheral fat stores and protein degradation from muscle increases circulating free FAs and amino acids to provide substrates for hepatic glucose synthesis[174]. However, when cortisol levels are chronically elevated (hypercortisolism), body fat distribution is disturbed (increased visceral adiposity), insulin signaling decays, and metabolic abnormalities arise, resulting in MetS[175]. Hypercortisolism is a condition characteristic of Cushing’s syndrome. Individuals with this syndrome show increased and abnormal distribution of fatty mass (central obesity), hypertension, and IR. In addition, they are prone to develop NAFLD due to fat accumulation in liver and adipose tissue and decreased muscle mass (sarcopenia) by GCs’ stimulation of protein breakdown and inhibition of protein synthesis[176].
GCs are employed in numerous autoimmune and inflammatory diseases due to their anti-inflammatory and immunosuppressive capacities. Long-term administration of a high dose of some GCs is frequently associated with hyperglycemia, fat mass redistribution, and IR, effects similar to those seen in Cushing’s syndrome. High-dose long-term use of glucocorticoids can induce painless myopathy that usually resolves after treatment withdrawal and exercise[177]. Moreover, fatty liver development also represents a typical side-effect of long-term systemic glucocorticoids treatment during anti-inflammatory therapy that may contribute to hepatic steatosis[178].
Dysregulation of adipose tissue physiology is one of the primary events in the development of IR syndrome[179]. Skeletal muscle and liver also play an important role given their characteristic response to insulin[38,180]. In liver and adipose tissue, the ability of GCs to bind and activate the GR is controlled by
The relationship between the 11β-HSD1 function and metabolic disorders has been well established in murine models. Deletion of 11β-HSD1 protects against GC-induced hepatic IR and skeletal muscle myopathy. In contrast, hepatic over-expression of this enzyme increases liver TG content, supporting the notion of the emergence of an adverse metabolic profile in the presence of elevated cortisol[182,183]. Additionally, pharmacological inhibition of 11β-HSD1 protects against high-fat diet-induced hepatic steatosis by inhibiting lipolysis in adipose tissues and lipogenesis in the liver[184]. 11β-HSD1 antisense oligonucleotide treatment protected C57BL/6J mice from hepatic steatosis induced by a Western-type diet by decreasing de novo lipogenesis and increasing β-oxidation[185]. In humans, 11β-HSD1 gene expression does not seem to be involved in the pathogenesis of fatty liver or NASH[186]. However, in subjects with hepatic steatosis, there is induction of hepatic 11β-HSD1 expression and activity that enhances hepatic cortisol levels, apparently due to the inflammatory response present in NASH[187]. In obese individuals, overexpression of 11β-HSD1 in visceral adipose tissue but not in the liver is associated with the presence of NAFLD[188].
A growing body of evidence appears to implicate GCs in the development and progression of NAFLD. Patients with NAFLD show overactivity of the HPA axis, leading to a subclinical hypercortisolism state closely associated with the severity of the liver histopathology[189]. Additionally, a patient with autonomous cortisol secretion due to an adrenal adenoma showed biochemical remission of NAFLD when treated with mifepristone (a competitive GR antagonist). The authors pointed out that autonomous cortisol secretion from adrenal adenomas could contribute to metabolic and liver abnormalities in patients with NAFLD[190]. GCs enhance adipocyte lipolysis through an increased expression of lipases and a decrease in glucose uptake by downregulating GLUT1 and GLUT4 transporters, which promote IR via a decrease in IRS1 expression and activity. As a consequence of adipose tissue IR, the lipids originating from the adipose tissue are re-esterified and accumulated in the liver. The mechanisms allowing GCs to switch from glucose storage to hepatic glucose production in an acute stressful situation are similar to those promoting hepatic IR by counteracting the effects of insulin on the liver. In the liver, the activation of the GC-GR signaling pathway inhibits the insulin receptor pathway and Akt activity and induces FOXO1, which in turn stimulates phosphoenolpyruvate carboxykinase and glucose-6-phosphatase expression and, ultimately, glucose production[173,191].
At the muscle level, hypercortisolemia disrupts the metabolism and alters mitochondrial functions. The decrease of skeletal muscle mass, associated with fat accumulation within the muscle, drives sarcopenia and myosteatosis development[192]. Skeletal muscle mass decreases as a result of both increased proteolysis and decreased protein synthesis. Cortisol activates the ubiquitin-proteasome and autophagy lysosomal systems through the increased expression of genes involved in muscular atrophy, namely FOXO transcription factors family, Atrogin-1, and MuRF-1 (muscle-specific E3 ubiquitin ligases). Inhibition of muscle protein synthesis results mainly from the GC-induced reduction of the mammalian target of rapamycin (mTOR) signaling pathway[193,194]. GCs also exacerbate the loss of protein and muscle mass by inhibiting IGF-1 and increasing myostatin expression[195]. Myostatin negatively regulates AKT/mTOR signaling pathway, which controls protein synthesis and is activated by insulin and IGF-1. Factors that modulate the insulin/IGF-1 signaling pathway are potential regulators of the catabolic effects of GCs[196]. Thus, myostatin is involved in the effect of GCs on muscle atrophy, reducing the IGF-1/PI3K/AKT signaling pathway by inhibiting AKT phosphorylation[197].
Mildly elevated cortisol concentration due to chronic stress is becoming more frequent in the general population, with the consequent risk of developing hepatic steatosis and muscle wasting. Mood disorders seem related to NAFLD and sarcopenia. NAFLD is significantly associated with depression and severe NAFLD with anxiety compared to non-NAFLD in women[198]. Another study performed in patients with NAFLD demonstrated that depression is associated with more severe liver histological abnormalities (hepatocyte ballooning)[199]. A relationship between psychological health and sarcopenia also has been recognized. In a cross-sectional observational study in China, diagnostic features of sarcopenia such as strength of the leg muscles (chair stand test) and physical performance (gait speed and standing balance test) are associated with depressive mood[200]. These pieces of evidence indicate that anxiety and depressive symptoms alter the functioning of the HPA axis, which in turn might contribute to the development of diseases associated with chronic hypercortisolism.
CONCLUSION
Sarcopenia is a muscle-related disorder characterized by the gradual loss of muscle mass and function, while NAFLD is the hepatic accumulation of lipids that leads to end-stage liver problems. Improving diet quality and an increase in physical activity reduce the risk of developing NAFLD and improve skeletal muscle functional capacity on sarcopenia. Sarcopenia and NAFLD, although being different disorders, are significantly correlated, and their progression seems to occur in a mutually synergistic fashion, leading to augmented adverse outcomes. However, it is not yet known if sarcopenia is the cause or consequence of NAFLD progression. These diseases share pathophysiological drivers. Ageing, chronic inflammation, obesity, and dyslipidemia promote insulin resistance that constitutes the principal driver of these disorders. Biochemical liver-muscle crosstalks, through various signaling pathways modulated by insulin resistance, lead to the progression of both disorders. Cortisol is strongly associated with insulin sensitivity and controls it negatively, providing an essential role in the muscle-liver axis. Hypercortisolism has been related to the development of NAFLD and sarcopenia, demonstrating that increased cortisol is a risk factor for either disease. Mood disorders are among the factors that lead to hypercortisolism and could be a feature to consider for the diagnosis of sarcopenia and NAFLD.
DECLARATIONS
Authors’ contributionsDrafting and review of the manuscript: Arrese M, Cabello-Verrugio C, Arab JP, Barrera F, Baudrand R, Guarda FJ, Gul I, Cabrera D
Figures design and drawing: Cabello-Verrugio C, Cabrera D
Original ideas and proposed hypothesis: Arrese M, Baudrand R, Cabrera D
All authors have read and approved the final version of the manuscript.
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was funded by grants from National Fund for Science and Technological Development (FONDECYT #1150327 and #1191145 to Arrese M; #1200227 to Arab JP; #1190419 to Baudrand R and #1191183 to Barrera F; #1211879 to Cabrera D; #1200944 to Cabello-Verrugio C); Millennium Institute on Immunology and Immunotherapy [P09-016-F (Cabello-Verrugio C)], Basal Grant CEDENNA [AFB180001 (Cabello-Verrugio C)], the Comisión Nacional de Investigación Científica y Tecnológica (CONICYT, AFB170005, CARE Chile UC)], and ANID-Anillo de Investigación en Ciencia y Tecnología [ACT210039 (Baudrand R)].
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethics approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
2. Siparsky PN, Kirkendall DT, Garrett WE Jr. Muscle changes in aging: understanding sarcopenia. Sports Health 2014;6:36-40.
3. Cruz-Jentoft AJ, Bahat G, Bauer J, et al. Writing Group for the European Working Group on Sarcopenia in Older People 2 (EWGSOP2). Sarcopenia: revised European consensus on definition and diagnosis. Age Ageing 2019;48:16-31.
4. Chakravarthy MV, Siddiqui MS, Forsgren MF, Sanyal AJ. Harnessing muscle-liver crosstalk to treat nonalcoholic steatohepatitis. Front Endocrinol (Lausanne) 2020;11:592373.
5. Hong HC, Hwang SY, Choi HY, et al. Relationship between sarcopenia and nonalcoholic fatty liver disease: the Korean Sarcopenic Obesity Study. Hepatology 2014;59:1772-8.
6. Koo BK, Kim D, Joo SK, et al. Sarcopenia is an independent risk factor for non-alcoholic steatohepatitis and significant fibrosis. J Hepatol 2017;66:123-31.
7. Cai C, Song X, Chen Y, Chen X, Yu C. Relationship between relative skeletal muscle mass and nonalcoholic fatty liver disease: a systematic review and meta-analysis. Hepatol Int 2020;14:115-26.
8. Kim G, Lee SE, Lee YB, et al. Relationship between relative skeletal muscle mass and nonalcoholic fatty liver disease: a 7-year longitudinal study. Hepatology 2018;68:1755-68.
9. Mikolasevic I, Pavic T, Kanizaj TF, Bender DV, Domislovic V, Krznaric Z. Nonalcoholic fatty liver disease and sarcopenia: where do we stand? Can J Gastroenterol Hepatol 2020;2020:8859719.
11. Younossi Z, Tacke F, Arrese M, et al. Global perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 2019;69:2672-82.
12. Su W, Wang Y, Jia X, et al. Comparative proteomic study reveals 17β-HSD13 as a pathogenic protein in nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A 2014;111:11437-42.
13. Altajar S, Baffy G. Skeletal muscle dysfunction in the development and progression of nonalcoholic fatty liver disease. J Clin Transl Hepatol 2020;8:414-23.
14. Severinsen MCK, Pedersen BK. Muscle-organ crosstalk: the emerging roles of myokines. Endocr Rev 2020;41:594-609.
15. de Oliveira Dos Santos AR, de Oliveira Zanuso B, Miola VFB, et al. Adipokines, myokines, and hepatokines: crosstalk and metabolic repercussions. Int J Mol Sci 2021;22:2639.
16. Pedersen BK, Febbraio MA. Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev Endocrinol 2012;8:457-65.
17. Keller K, Engelhardt M. Strength and muscle mass loss with aging process. Age and strength loss. Muscles Ligaments Tendons J 2014;3:346-50.
18. Beaudart C, McCloskey E, Bruyère O, et al. Sarcopenia in daily practice: assessment and management. BMC Geriatr 2016;16:170.
19. Shafiee G, Keshtkar A, Soltani A, Ahadi Z, Larijani B, Heshmat R. Prevalence of sarcopenia in the world: a systematic review and meta- analysis of general population studies. J Diabetes Metab Disord 2017;16:21.
20. Papadopoulou SK, Tsintavis P, Potsaki P, Papandreou D. Differences in the prevalence of sarcopenia in community-dwelling, nursing home and hospitalized individuals. A systematic review and meta-analysis. J Nutr Health Aging 2020;24:83-90.
21. Aleixo GFP, Williams GR, Nyrop KA, Muss HB, Shachar SS. Muscle composition and outcomes in patients with breast cancer: meta-analysis and systematic review. Breast Cancer Res Treat 2019;177:569-79.
22. Bone AE, Hepgul N, Kon S, Maddocks M. Sarcopenia and frailty in chronic respiratory disease. Chron Respir Dis 2017;14:85-99.
23. Sui SX, Williams LJ, Holloway-Kew KL, Hyde NK, Pasco JA. Skeletal muscle health and cognitive function: a narrative review. Int J Mol Sci 2020;22:255.
24. Petta S, Ciminnisi S, Di Marco V, et al. Sarcopenia is associated with severe liver fibrosis in patients with non-alcoholic fatty liver disease. Aliment Pharmacol Ther 2017;45:510-8.
25. Wilkinson TJ, Miksza J, Yates T, et al. Association of sarcopenia with mortality and end-stage renal disease in those with chronic kidney disease: a UK Biobank study. J Cachexia Sarcopenia Muscle 2021;12:586-98.
26. Vogel F, Braun LT, Rubinstein G, et al. Persisting muscle dysfunction in Cushing’s syndrome despite biochemical remission. J Clin Endocrinol Metab 2020;105:e4490-8.
27. Fuggle N, Shaw S, Dennison E, Cooper C. Sarcopenia. Best Pract Res Clin Rheumatol 2017;31:218-42.
28. Riuzzi F, Sorci G, Arcuri C, et al. Cellular and molecular mechanisms of sarcopenia: the S100B perspective. J Cachexia Sarcopenia Muscle 2018;9:1255-68.
29. Dalle S, Rossmeislova L, Koppo K. The role of inflammation in age-related sarcopenia. Front Physiol 2017;8:1045.
30. Alemán H, Esparza J, Ramirez FA, Astiazaran H, Payette H. Longitudinal evidence on the association between interleukin-6 and C-reactive protein with the loss of total appendicular skeletal muscle in free-living older men and women. Age Ageing 2011;40:469-75.
31. Li CW, Yu K, Shyh-Chang N, et al. Circulating factors associated with sarcopenia during ageing and after intensive lifestyle intervention. J Cachexia Sarcopenia Muscle 2019;10:586-600.
32. Sakellariou GK, Lightfoot AP, Earl KE, Stofanko M, McDonagh B. Redox homeostasis and age-related deficits in neuromuscular integrity and function. J Cachexia Sarcopenia Muscle 2017;8:881-906.
33. Thoma A, Akter-Miah T, Reade RL, Lightfoot AP. Targeting reactive oxygen species (ROS) to combat the age-related loss of muscle mass and function. Biogerontology 2020;21:475-84.
34. Boengler K, Kosiol M, Mayr M, Schulz R, Rohrbach S. Mitochondria and ageing: role in heart, skeletal muscle and adipose tissue. J Cachexia Sarcopenia Muscle 2017;8:349-69.
35. Dirks AJ, Leeuwenburgh C. The role of apoptosis in age-related skeletal muscle atrophy. Sports Med 2005;35:473-83.
36. Lo JH, U KP, Yiu T, Ong MT, Lee WY. Sarcopenia: current treatments and new regenerative therapeutic approaches. J Orthop Translat 2020;23:38-52.
37. Hunter GR, Singh H, Carter SJ, Bryan DR, Fisher G. Sarcopenia and Its Implications for Metabolic Health. J Obes 2019;2019:8031705.
38. Cleasby ME, Jamieson PM, Atherton PJ. Insulin resistance and sarcopenia: mechanistic links between common co-morbidities. J Endocrinol 2016;229:R67-81.
39. Schoufour JD, Tieland M, Barazzoni R, et al. The relevance of diet, physical activity, exercise, and persuasive technology in the prevention and treatment of sarcopenic obesity in older adults. Front Nutr 2021;8:661449.
40. Guillet C, Boirie Y. Insulin resistance: a contributing factor to age-related muscle mass loss? Diabetes Metab 2005;31:5S20-6.
41. Katsanos CS, Mandarino LJ. Protein metabolism in human obesity: a shift in focus from whole-body to skeletal muscle. Obesity (Silver Spring) 2011;19:469-75.
42. Borst SE. Interventions for sarcopenia and muscle weakness in older people. Age Ageing 2004;33:548-55.
43. Christiansen AR, Lipshultz LI, Hotaling JM, Pastuszak AW. Selective androgen receptor modulators: the future of androgen therapy? Transl Androl Urol 2020;9:S135-48.
44. Neil D, Clark RV, Magee M, et al. GSK2881078, a SARM, Produces dose-dependent increases in lean mass in healthy older men and women. J Clin Endocrinol Metab 2018;103:3215-24.
45. Dalton JT, Barnette KG, Bohl CE, et al. The selective androgen receptor modulator GTx-024 (enobosarm) improves lean body mass and physical function in healthy elderly men and postmenopausal women: results of a double-blind, placebo-controlled phase II trial. J Cachexia Sarcopenia Muscle 2011;2:153-61.
46. Dobs AS, Boccia RV, Croot CC, et al. Effects of enobosarm on muscle wasting and physical function in patients with cancer: a double-blind, randomised controlled phase 2 trial. Lancet Oncol 2013;14:335-45.
47. Lambert CP. Should the FDA’s criteria for the clinical efficacy of cachexia drugs be changed? J Cachexia Sarcopenia Muscle 2021;12:531-2.
48. Elkina Y, von Haehling S, Anker SD, Springer J. The role of myostatin in muscle wasting: an overview. J Cachexia Sarcopenia Muscle 2011;2:143-51.
49. Suh J, Lee YS. Myostatin inhibitors: panacea or predicament for musculoskeletal disorders? J Bone Metab 2020;27:151-65.
50. Consitt LA, Clark BC. The vicious cycle of myostatin signaling in sarcopenic obesity: myostatin role in skeletal muscle growth, insulin signaling and implications for clinical trials. J Frailty Aging 2018;7:21-7.
51. Guo T, Jou W, Chanturiya T, Portas J, Gavrilova O, McPherron AC. Myostatin inhibition in muscle, but not adipose tissue, decreases fat mass and improves insulin sensitivity. PLoS One 2009;4:e4937.
52. Dong J, Dong Y, Dong Y, Chen F, Mitch WE, Zhang L. Inhibition of myostatin in mice improves insulin sensitivity via irisin-mediated cross talk between muscle and adipose tissues. Int J Obes (Lond) 2016;40:434-42.
53. Zhu HJ, Yang HB, Wang XQ, et al. Serum myostatin levels are elevated in overweight patients. TOOBESJ 2014;6:31-7.
54. Milan G, Dalla Nora E, Pilon C, et al. Changes in muscle myostatin expression in obese subjects after weight loss. J Clin Endocrinol Metab 2004;89:2724-7.
55. Pan H, Ping XC, Zhu HJ, et al. Association of myostatin gene polymorphisms with obesity in Chinese north Han human subjects. Gene 2012;494:237-41.
56. Hittel DS, Axelson M, Sarna N, Shearer J, Huffman KM, Kraus WE. Myostatin decreases with aerobic exercise and associates with insulin resistance. Med Sci Sports Exerc 2010;42:2023-9.
57. Saitoh M, Ishida J, Ebner N, Anker SD, Von Haehling S. Myostatin inhibitors as pharmacological treatment for muscle wasting and muscular dystrophy. JCSM Clinical Reports 2017;2:1-10.
58. Garito T, Roubenoff R, Hompesch M, et al. Bimagrumab improves body composition and insulin sensitivity in insulin-resistant individuals. Diabetes Obes Metab 2018;20:94-102.
59. Rooks D, Petricoul O, Praestgaard J, Bartlett M, Laurent D, Roubenoff R. Safety and pharmacokinetics of bimagrumab in healthy older and obese adults with body composition changes in the older cohort. J Cachexia Sarcopenia Muscle 2020;11:1525-34.
60. Witham MD, Sumukadas D, McMurdo ME. ACE inhibitors for sarcopenia--as good as exercise training? Age Ageing 2008;37:363-5.
61. Ata AM, Kara M, Ekiz T, et al. Reassessing sarcopenia in hypertension: STAR and ACE inhibitors excel. Int J Clin Pract 2021;75:e13800.
62. Zhou LS, Xu LJ, Wang XQ, Huang YH, Xiao Q. Effect of angiotensin-converting enzyme inhibitors on physical function in elderly subjects: a systematic review and meta-analysis. Drugs Aging 2015;32:727-35.
63. Caulfield L, Heslop P, Walesby KE, Sumukadas D, Sayer AA, Witham MD. Effect of angiotensin system inhibitors on physical performance in older people - a systematic review and meta-analysis. J Am Med Dir Assoc 2021;22:1215-21.e2.
64. Papadopoulou SK. Sarcopenia: a contemporary health problem among older adult populations. Nutrients 2020;12:1293.
65. Hardee JP, Lynch GS. Current pharmacotherapies for sarcopenia. Expert Opin Pharmacother 2019;20:1645-57.
66. Seene T, Kaasik P. Muscle weakness in the elderly: role of sarcopenia, dynapenia, and possibilities for rehabilitation. Eur Rev Aging Phys Act 2012;9:109-17.
67. Nicklas BJ, Hsu FC, Brinkley TJ, et al. Exercise training and plasma C-reactive protein and interleukin-6 in elderly people. J Am Geriatr Soc 2008;56:2045-52.
68. Starkie R, Ostrowski SR, Jauffred S, Febbraio M, Pedersen BK. Exercise and IL-6 infusion inhibit endotoxin-induced TNF-alpha production in humans. FASEB J 2003;17:884-6.
69. Lang T, Streeper T, Cawthon P, Baldwin K, Taaffe DR, Harris TB. Sarcopenia: etiology, clinical consequences, intervention, and assessment. Osteoporos Int 2010;21:543-59.
70. Phillips SM. Nutritional supplements in support of resistance exercise to counter age-related sarcopenia. Adv Nutr 2015;6:452-60.
71. Tessier AJ, Chevalier S. An update on protein, leucine, Omega-3 fatty acids, and vitamin D in the prevention and treatment of sarcopenia and functional decline. Nutrients 2018;10:1099.
72. Coelho-Júnior HJ, Milano-Teixeira L, Rodrigues B, Bacurau R, Marzetti E, Uchida M. Relative protein intake and physical function in older adults: a systematic review and meta-analysis of observational studies. Nutrients 2018;10:1330.
73. Kirk B, Mooney K, Amirabdollahian F, Khaiyat O. Exercise and dietary-protein as a countermeasure to skeletal muscle weakness: Liverpool Hope University - Sarcopenia Aging Trial (LHU-SAT). Front Physiol 2019;10:445.
74. Kwak JY, Kwon KS. Pharmacological interventions for treatment of sarcopenia: current status of drug development for sarcopenia. Ann Geriatr Med Res 2019;23:98-104.
75. Rondanelli M, Miccono A, Peroni G, et al. A systematic review on the effects of botanicals on skeletal muscle health in order to prevent sarcopenia. Evid Based Complement Alternat Med 2016;2016:5970367.
76. Wei Y, Chen Y, Qiu Y, et al. Prevention of muscle wasting by CRISPR/Cas9-mediated disruption of myostatin in vivo. Mol Ther 2016;24:1889-91.
77. Fan J, Kou X, Yang Y, Chen N. MicroRNA-regulated proinflammatory cytokines in sarcopenia. Mediators Inflamm 2016;2016:1438686.
78. Tompkins BA, DiFede DL, Khan A, et al. Allogeneic mesenchymal stem cells ameliorate aging frailty: a phase II randomized, double-blind, placebo-controlled clinical trial. J Gerontol A Biol Sci Med Sci 2017;72:1513-22.
79. Zhu Y, Ge J, Huang C, Liu H, Jiang H. Application of mesenchymal stem cell therapy for aging frailty: from mechanisms to therapeutics. Theranostics 2021;11:5675-85.
80. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016;64:73-84.
82. Scapaticci S, D’Adamo E, Mohn A, Chiarelli F, Giannini C. Non-alcoholic fatty liver disease in obese youth with insulin resistance and type 2 diabetes. Front Endocrinol (Lausanne) 2021;12:639548.
83. Eslam M, Sanyal AJ, George J. International Consensus Panel. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 2020;158:1999-2014.e1.
84. Association for the Study of the Liver (EASL), European Association for the Study of Diabetes (EASD), European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J Hepatol 2016;64:1388-402.
85. Stefan N, Häring H, Cusi K. Non-alcoholic fatty liver disease: causes, diagnosis, cardiometabolic consequences, and treatment strategies. Lancet Diabetes Endocrinol 2019;7:313-24.
86. Clark JM, Brancati FL, Diehl AM. Nonalcoholic fatty liver disease. Gastroenterology 2002;122:1649-57.
87. Pydyn N, Miękus K, Jura J, Kotlinowski J. New therapeutic strategies in nonalcoholic fatty liver disease: a focus on promising drugs for nonalcoholic steatohepatitis. Pharmacol Rep 2020;72:1-12.
88. Pais R, Charlotte F, Fedchuk L, et al. LIDO Study Group. A systematic review of follow-up biopsies reveals disease progression in patients with non-alcoholic fatty liver. J Hepatol 2013;59:550-6.
89. Angulo P, Kleiner DE, Dam-Larsen S, et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015;149:389-97.e10.
90. Ekstedt M, Hagström H, Nasr P, et al. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015;61:1547-54.
91. Kanwal F, Kramer JR, Mapakshi S, et al. Risk of hepatocellular cancer in patients with non-alcoholic fatty liver disease. Gastroenterology 2018;155:1828-37.e2.
92. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med 2018;24:908-22.
93. Sanyal AJ. Past, present and future perspectives in nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol 2019;16:377-86.
94. Juanola O, Martínez-López S, Francés R, Gómez-Hurtado I. Non-alcoholic fatty liver disease: metabolic, genetic, epigenetic and environmental risk factors. Int J Environ Res Public Health 2021;18:5227.
95. Eslam M, George J. Genetic contributions to NAFLD: leveraging shared genetics to uncover systems biology. Nat Rev Gastroenterol Hepatol 2020;17:40-52.
96. Eslam M, Valenti L, Romeo S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J Hepatol 2018;68:268-79.
97. Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461-5.
98. Unalp-Arida A, Ruhl CE. Patatin-like phospholipase domain-containing protein 3 I148M and liver fat and fibrosis scores predict liver disease mortality in the U.S. population. Hepatology 2020;71:820-34.
99. Ma J, Hennein R, Liu C, et al. Improved diet quality associates with reduction in liver fat, particularly in individuals with high genetic risk scores for nonalcoholic fatty liver disease. Gastroenterology 2018;155:107-17.
100. Sung KC, Ryu S, Lee JY, Kim JY, Wild SH, Byrne CD. Effect of exercise on the development of new fatty liver and the resolution of existing fatty liver. J Hepatol 2016;65:791-7.
101. Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology 2015;149:367-78.e5; quiz e14-5.
102. Veena J, Muragundla A, Sidgiddi S, Subramaniam S. Non-alcoholic fatty liver disease: need for a balanced nutritional source. Br J Nutr 2014;112:1858-72.
103. DeFronzo RA, Tripathy D, Schwenke DC, et al. ACT NOW Study. Pioglitazone for diabetes prevention in impaired glucose tolerance. N Engl J Med 2011;364:1104-15.
104. Neuschwander-Tetri BA. Therapeutic landscape for NAFLD in 2020. Gastroenterology 2020;158:1984-98.e3.
105. Gan D, Wang L, Jia M, et al. Low muscle mass and low muscle strength associate with nonalcoholic fatty liver disease. Clin Nutr 2020;39:1124-30.
106. Hashimoto Y, Osaka T, Fukuda T, Tanaka M, Yamazaki M, Fukui M. The relationship between hepatic steatosis and skeletal muscle mass index in men with type 2 diabetes. Endocr J 2016;63:877-84.
107. Lee YH, Kim SU, Song K, et al. Sarcopenia is associated with significant liver fibrosis independently of obesity and insulin resistance in nonalcoholic fatty liver disease: Nationwide surveys (KNHANES 2008-2011). Hepatology 2016;63:776-86.
108. Wijarnpreecha K, Kim D, Raymond P, Scribani M, Ahmed A. Associations between sarcopenia and nonalcoholic fatty liver disease and advanced fibrosis in the USA. Eur J Gastroenterol Hepatol 2019;31:1121-8.
109. Montano-Loza AJ, Angulo P, Meza-Junco J, et al. Sarcopenic obesity and myosteatosis are associated with higher mortality in patients with cirrhosis. J Cachexia Sarcopenia Muscle 2016;7:126-35.
110. Sherif O, Dhaliwal A, Newsome PN, Armstrong MJ. Sarcopenia in nonalcoholic fatty liver disease: new challenges for clinical practice. Expert Rev Gastroenterol Hepatol 2020;14:197-205.
111. Okamura T, Hashimoto Y, Hamaguchi M, Obora A, Kojima T, Fukui M. The visceral adiposity index is a predictor of incident nonalcoholic fatty liver disease: a population-based longitudinal study. Clin Res Hepatol Gastroenterol 2020;44:375-83.
112. Mirza MS. Obesity, visceral fat, and NAFLD: querying the role of adipokines in the progression of nonalcoholic fatty liver disease. ISRN Gastroenterol 2011;2011:592404.
113. Petta S, Amato MC, Di Marco V, et al. Visceral adiposity index is associated with significant fibrosis in patients with non-alcoholic fatty liver disease. Aliment Pharmacol Ther 2012;35:238-47.
114. Ismaiel A, Jaaouani A, Leucuta DC, Popa SL, Dumitrascu DL. The visceral adiposity index in non-alcoholic fatty liver disease and liver fibrosis-systematic review and meta-analysis. Biomedicines 2021;9:1890.
115. Andrade LJ, Melo PR, Paraná R, Daltro C. Grading scale of visceral adipose tissue thickness and their relation to the nonalcoholic fatty liver disease. Arq Gastroenterol 2014;51:118-22.
116. Francque S, Verrijken A, Mertens I, et al. Visceral adiposity and insulin resistance are independent predictors of the presence of non-cirrhotic NAFLD-related portal hypertension. Int J Obes (Lond) 2011;35:270-8.
117. Lee S, Kim KW, Lee J, et al. Visceral adiposity as a risk factor for lean non-alcoholic fatty liver disease in potential living liver donors. J Gastroenterol Hepatol 2021;36:3212-8.
118. Xu C, Ma Z, Wang Y, et al. Visceral adiposity index as a predictor of NAFLD: a prospective study with 4-year follow-up. Liver Int 2018;38:2294-300.
119. Kure T, Mawatari S, Imamura Y, et al. Nonalcoholic fatty liver disease is associated with both subcutaneous and visceral adiposity: a cross-sectional study. Medicine (Baltimore) 2019;98:e17879.
120. Vural Keskinler M, Mutlu HH, Sirin A, et al. Visceral adiposity index as a practical tool in patients with biopsy-proven nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Metab Syndr Relat Disord 2021;19:26-31.
121. Nachit M, Kwanten WJ, Thissen JP, et al. Muscle fat content is strongly associated with NASH: A longitudinal study in patients with morbid obesity. J Hepatol 2021;75:292-301.
122. De Munck TJI, Verhaegh P, Lodewick T, et al. Myosteatosis in nonalcoholic fatty liver disease: an exploratory study. Clin Res Hepatol Gastroenterol 2021;45:101500.
123. Nachit M, De Rudder M, Thissen JP, et al. Myosteatosis rather than sarcopenia associates with non-alcoholic steatohepatitis in non-alcoholic fatty liver disease preclinical models. J Cachexia Sarcopenia Muscle 2021;12:144-58.
124. Linge J, Ekstedt M, Dahlqvist Leinhard O. Adverse muscle composition is linked to poor functional performance and metabolic comorbidities in NAFLD. JHEP Rep 2021;3:100197.
125. Nachit M, Lanthier N, Rodriguez J, et al. A dynamic association between myosteatosis and liver stiffness: results from a prospective interventional study in obese patients. JHEP Rep 2021;3:100323.
126. Nachit M, Lanthier N, Rodriguez J, et al. An unsuspected link between myosteatosis and NAFLD grading in a prospective nutritional intervention study in obese patients. Hepatology 2020;72:891A-2A.
127. Ebadi M, Montano-Loza AJ. Clinical relevance of skeletal muscle abnormalities in patients with cirrhosis. Dig Liver Dis 2019;51:1493-9.
128. Hsieh YC, Joo SK, Koo BK, Lin HC, Kim W. Muscle alterations are independently associated with significant fibrosis in patients with nonalcoholic fatty liver disease. Liver Int 2021;41:494-504.
130. Jensen-Cody SO, Potthoff MJ. Hepatokines and metabolism: deciphering communication from the liver. Mol Metab 2021;44:101138.
131. Chadt A, Al-Hasani H. Glucose transporters in adipose tissue, liver, and skeletal muscle in metabolic health and disease. Pflugers Arch 2020;472:1273-98.
132. Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest 2008;118:829-38.
133. Tilg H, Moschen AR. Insulin resistance, inflammation, and non-alcoholic fatty liver disease. Trends Endocrinol Metab 2008;19:371-9.
134. Brown MS, Goldstein JL. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab 2008;7:95-6.
135. Ipsen DH, Lykkesfeldt J, Tveden-Nyborg P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol Life Sci 2018;75:3313-27.
136. Peverill W, Powell LW, Skoien R. Evolving concepts in the pathogenesis of NASH: beyond steatosis and inflammation. Int J Mol Sci 2014;15:8591-638.
137. Cusi K. Role of insulin resistance and lipotoxicity in non-alcoholic steatohepatitis. Clin Liver Dis 2009;13:545-63.
138. Sabio G, Das M, Mora A, et al. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 2008;322:1539-43.
139. Coulon S, Francque S, Colle I, et al. Evaluation of inflammatory and angiogenic factors in patients with non-alcoholic fatty liver disease. Cytokine 2012;59:442-9.
140. Lehrskov LL, Christensen RH. The role of interleukin-6 in glucose homeostasis and lipid metabolism. Semin Immunopathol 2019;41:491-9.
141. Sakurai Y, Kubota N, Yamauchi T, Kadowaki T. Role of insulin resistance in MAFLD. Int J Mol Sci 2021;22:4156.
142. Zhang L, Keung W, Samokhvalov V, Wang W, Lopaschuk GD. Role of fatty acid uptake and fatty acid beta-oxidation in mediating insulin resistance in heart and skeletal muscle. Biochim Biophys Acta 2010;1801:1-22.
143. Hegarty BD, Furler SM, Ye J, Cooney GJ, Kraegen EW. The role of intramuscular lipid in insulin resistance. Acta Physiol Scand 2003;178:373-83.
144. Wu H, Ballantyne CM. Skeletal muscle inflammation and insulin resistance in obesity. J Clin Invest 2017;127:43-54.
145. Austin RL, Rune A, Bouzakri K, Zierath JR, Krook A. siRNA-mediated reduction of inhibitor of nuclear factor-kappaB kinase prevents tumor necrosis factor-alpha-induced insulin resistance in human skeletal muscle. Diabetes 2008;57:2066-73.
146. Aleffi S, Petrai I, Bertolani C, et al. Upregulation of proinflammatory and proangiogenic cytokines by leptin in human hepatic stellate cells. Hepatology 2005;42:1339-48.
147. Angulo P, Alba LM, Petrovic LM, Adams LA, Lindor KD, Jensen MD. Leptin, insulin resistance, and liver fibrosis in human nonalcoholic fatty liver disease. J Hepatol 2004;41:943-9.
148. Maeda N, Takahashi M, Funahashi T, et al. PPARgamma ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes 2001;50:2094-9.
150. Musso G, Gambino R, Biroli G, et al. Hypoadiponectinemia predicts the severity of hepatic fibrosis and pancreatic Beta-cell dysfunction in nondiabetic nonobese patients with nonalcoholic steatohepatitis. Am J Gastroenterol 2005;100:2438-46.
151. Kellerer M, Koch M, Metzinger E, Mushack J, Capp E, Häring HU. Leptin activates PI-3 kinase in C2C12 myotubes via janus kinase-2 (JAK-2) and insulin receptor substrate-2 (IRS-2) dependent pathways. Diabetologia 1997;40:1358-62.
152. Minokoshi Y, Kahn BB. Role of AMP-activated protein kinase in leptin-induced fatty acid oxidation in muscle. Biochem Soc Trans 2003;31:196-201.
153. Ceddia RB. Direct metabolic regulation in skeletal muscle and fat tissue by leptin: implications for glucose and fatty acids homeostasis. Int J Obes (Lond) 2005;29:1175-83.
154. Wang F, So KF, Xiao J, Wang H. Organ-organ communication: the liver’s perspective. Theranostics 2021;11:3317-30.
155. Rittig K, Thamer C, Haupt A, et al. High plasma fetuin-A is associated with increased carotid intima-media thickness in a middle-aged population. Atherosclerosis 2009;207:341-2.
156. Ix JH, Wassel CL, Kanaya AM, et al. Health ABC Study. Fetuin-A and incident diabetes mellitus in older persons. JAMA 2008;300:182-8.
157. Chattopadhyay M, Mukherjee S, Chatterjee SK, et al. Impairment of energy sensors, SIRT1 and AMPK, in lipid induced inflamed adipocyte is regulated by Fetuin A. Cell Signal 2018;42:67-76.
158. Hansen JS, Rutti S, Arous C, et al. Circulating follistatin is liver-derived and regulated by the glucagon-to-insulin ratio. J Clin Endocrinol Metab 2016;101:550-60.
159. Brown ML, Bonomi L, Ungerleider N, et al. Follistatin and follistatin like-3 differentially regulate adiposity and glucose homeostasis. Obesity (Silver Spring) 2011;19:1940-9.
160. Watt MJ, Miotto PM, De Nardo W, Montgomery MK. The liver as an endocrine organ-linking NAFLD and insulin resistance. Endocr Rev 2019;40:1367-93.
161. Jung TW, Chung YH, Kim HC, Abd El-Aty AM, Jeong JH. Hyperlipidemia-induced hepassocin in the liver contributes to insulin resistance in skeletal muscle. Mol Cell Endocrinol 2018;470:26-33.
162. Lan F, Misu H, Chikamoto K, et al. LECT2 functions as a hepatokine that links obesity to skeletal muscle insulin resistance. Diabetes 2014;63:1649-64.
163. Misu H, Takamura T, Takayama H, et al. A liver-derived secretory protein, selenoprotein P, causes insulin resistance. Cell Metab 2010;12:483-95.
164. Succurro E, Andreozzi F, Marini MA, et al. Low plasma insulin-like growth factor-1 levels are associated with reduced insulin sensitivity and increased insulin secretion in nondiabetic subjects. Nutr Metab Cardiovasc Dis 2009;19:713-9.
165. Wyk JJ, Smith EP. Insulin-like growth factors and skeletal growth: possibilities for therapeutic interventions. J Clin Endocrinol Metab 1999;84:4349-54.
166. Fernández AM, Kim JK, Yakar S, et al. Functional inactivation of the IGF-I and insulin receptors in skeletal muscle causes type 2 diabetes. Genes Dev 2001;15:1926-34.
167. Laron Z, Avitzur Y, Klinger B. Carbohydrate metabolism in primary growth hormone resistance (Laron syndrome) before and during insulin-like growth factor-I treatment. Metabolism 1995;44:113-8.
168. Arturi F, Succurro E, Procopio C, et al. Nonalcoholic fatty liver disease is associated with low circulating levels of insulin-like growth factor-I. J Clin Endocrinol Metab 2011;96:E1640-4.
169. Møller N, Gormsen LC, Schmitz O, Lund S, Jørgensen JO, Jessen N. Free fatty acids inhibit growth hormone/signal transducer and activator of transcription-5 signaling in human muscle: a potential feedback mechanism. J Clin Endocrinol Metab 2009;94:2204-7.
170. Wilkes JJ, Lloyd DJ, Gekakis N. Loss-of-function mutation in myostatin reduces tumor necrosis factor alpha production and protects liver against obesity-induced insulin resistance. Diabetes 2009;58:1133-43.
171. García PS, Cabbabe A, Kambadur R, Nicholas G, Csete M. Brief-reports: elevated myostatin levels in patients with liver disease: a potential contributor to skeletal muscle wasting. Anesth Analg 2010;111:707-9.
172. Nishikawa H, Enomoto H, Ishii A, et al. Elevated serum myostatin level is associated with worse survival in patients with liver cirrhosis. J Cachexia Sarcopenia Muscle 2017;8:915-25.
173. Beaupere C, Liboz A, Fève B, Blondeau B, Guillemain G. Molecular mechanisms of glucocorticoid-induced insulin resistance. Int J Mol Sci 2021;22:623.
174. Jeong IK. The role of cortisol in the pathogenesis of the metabolic syndrome. Diabetes Metab J 2012;36:207-10.
175. Roberge C, Carpentier AC, Langlois MF, et al. Adrenocortical dysregulation as a major player in insulin resistance and onset of obesity. Am J Physiol Endocrinol Metab 2007;293:E1465-78.
176. Ferraù F, Korbonits M. Metabolic comorbidities in Cushing’s syndrome. Eur J Endocrinol 2015;173:M133-57.
177. Yasir M, Goyal A, Bansal P, Sonthalia S. . Corticosteroid adverse effects. Treasure Island (FL): StatPearls Publishing; 2022.
178. Yang M, Ma F, Guan M. Role of steroid hormones in the pathogenesis of nonalcoholic fatty liver disease. Metabolites 2021;11:320.
179. Lewis GF, Carpentier A, Adeli K, Giacca A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr Rev 2002;23:201-29.
180. Birkenfeld AL, Shulman GI. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology 2014;59:713-23.
182. Morgan SA, Hassan-Smith ZK, Doig CL, Sherlock M, Stewart PM, Lavery GG. Glucocorticoids and 11β-HSD1 are major regulators of intramyocellular protein metabolism. J Endocrinol 2016;229:277-86.
183. Morton NM, Holmes MC, Fiévet C, et al. Improved lipid and lipoprotein profile, hepatic insulin sensitivity, and glucose tolerance in 11beta-hydroxysteroid dehydrogenase type 1 null mice. J Biol Chem 2001;276:41293-300.
184. Li H, Sheng J, Wang J, et al. Selective inhibition of 11β-hydroxysteroid dehydrogenase type 1 attenuates high-fat diet-induced hepatic steatosis in mice. Drug Des Devel Ther 2021;15:2309-24.
185. Li G, Hernandez-Ono A, Crooke RM, Graham MJ, Ginsberg HN. Effects of antisense-mediated inhibition of 11β-hydroxysteroid dehydrogenase type 1 on hepatic lipid metabolism. J Lipid Res 2011;52:971-81.
186. Konopelska S, Kienitz T, Hughes B, et al. Hepatic 11beta-HSD1 mRNA expression in fatty liver and nonalcoholic steatohepatitis. Clin Endocrinol (Oxf) 2009;70:554-60.
187. Ahmed A, Rabbitt E, Brady T, et al. A switch in hepatic cortisol metabolism across the spectrum of non alcoholic fatty liver disease. PLoS One 2012;7:e29531.
188. Candia R, Riquelme A, Baudrand R, et al. Overexpression of 11β-hydroxysteroid dehydrogenase type 1 in visceral adipose tissue and portal hypercortisolism in non-alcoholic fatty liver disease. Liver Int 2012;32:392-9.
189. Targher G, Bertolini L, Rodella S, Zoppini G, Zenari L, Falezza G. Associations between liver histology and cortisol secretion in subjects with nonalcoholic fatty liver disease. Clin Endocrinol (Oxf) 2006;64:337-41.
190. Ragucci E, Nguyen D, Lamerson M, Moraitis AG. Effects of mifepristone on nonalcoholic fatty liver disease in a patient with a cortisol-secreting adrenal adenoma. Case Rep Endocrinol 2017;2017:6161348.
191. Rahimi L, Rajpal A, Ismail-Beigi F. Glucocorticoid-induced fatty liver disease. Diabetes Metab Syndr Obes 2020;13:1133-45.
192. Stefanaki C, Pervanidou P, Boschiero D, Chrousos GP. Chronic stress and body composition disorders: implications for health and disease. Hormones (Athens) 2018;17:33-43.
193. Schakman O, Kalista S, Barbé C, Loumaye A, Thissen JP. Glucocorticoid-induced skeletal muscle atrophy. Int J Biochem Cell Biol 2013;45:2163-72.
194. Kuo T, Harris CA, Wang JC. Metabolic functions of glucocorticoid receptor in skeletal muscle. Mol Cell Endocrinol 2013;380:79-88.
195. Schakman O, Gilson H, Thissen JP. Mechanisms of glucocorticoid-induced myopathy. J Endocrinol 2008;197:1-10.
196. Amirouche A, Durieux AC, Banzet S, et al. Down-regulation of Akt/mammalian target of rapamycin signaling pathway in response to myostatin overexpression in skeletal muscle. Endocrinology 2009;150:286-94.
197. McFarlane C, Plummer E, Thomas M, et al. Myostatin induces cachexia by activating the ubiquitin proteolytic system through an NF-kappaB-independent, FoxO1-dependent mechanism. J Cell Physiol 2006;209:501-14.
198. Choi JM, Chung GE, Kang SJ, et al. Association between anxiety and depression and nonalcoholic fatty liver disease. Front Med (Lausanne) 2020;7:585618.
199. Youssef NA, Abdelmalek MF, Binks M, et al. Associations of depression, anxiety and antidepressants with histological severity of nonalcoholic fatty liver disease. Liver Int 2013;33:1062-70.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Arrese M, Cabello-Verrugio C, Arab JP, Barrera F, Baudrand R, Guarda FJ, Gul I, Cabrera D. Sarcopenia in the setting of nonalcoholic fatty liver. Metab Target Organ Damage 2022;2:2. http://dx.doi.org/10.20517/mtod.2021.16
AMA Style
Arrese M, Cabello-Verrugio C, Arab JP, Barrera F, Baudrand R, Guarda FJ, Gul I, Cabrera D. Sarcopenia in the setting of nonalcoholic fatty liver. Metabolism and Target Organ Damage. 2022; 2(1): 2. http://dx.doi.org/10.20517/mtod.2021.16
Chicago/Turabian Style
Arrese, Marco, Claudio Cabello-Verrugio, Juan Pablo Arab, Francisco Barrera, Rene Baudrand, Francisco J. Guarda, Iram Gul, Daniel Cabrera. 2022. "Sarcopenia in the setting of nonalcoholic fatty liver" Metabolism and Target Organ Damage. 2, no.1: 2. http://dx.doi.org/10.20517/mtod.2021.16
ACS Style
Arrese, M.; Cabello-Verrugio C.; Arab JP.; Barrera F.; Baudrand R.; Guarda FJ.; Gul I.; Cabrera D. Sarcopenia in the setting of nonalcoholic fatty liver. Metab Target Organ Damage. 2022, 2, 2. http://dx.doi.org/10.20517/mtod.2021.16
About This Article
Copyright

Data & Comments
Data


 Cite This Article 44 clicks
Cite This Article 44 clicks

 Like This Article 30
likes
Like This Article 30
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.