
Arresting type 1 diabetes: are we there yet? Obstacles and opportunities

Abstract
More than 100 years after the discovery of insulin, the exact etiology and pathophysiology of type 1 diabetes (T1D) remains elusive, but our knowledge is growing. This leads to louder calls to initiate a risk screening for T1D in the general population. This risk screening could be based on the genetic risk (in the general population or targeted HLA genotyping in family members of persons with T1D) or on the screening for autoantibodies in blood (e.g., antibodies against insulin, GAD, IA2, or ZnT8). The presence of autoantibodies is known to convey a clearly increased risk of progressing to T1D, particularly when two or more antibody types are present. It remains a point of discussion whether screening efforts are cost-effective. At present, in the absence of interventions capable of delaying the onset of disease, the only benefit of screening is the earlier diagnosis of T1D, thus avoiding life-threatening diabetic ketoacidosis (DKA). Nevertheless, large consortia (e.g., INNODIA and TrialNet) are currently focusing on not only disease biomarkers but also biomarkers of therapeutic effect of interventions. All hope is thus focused on the arrival of intervention strategies that could arrest the ongoing immune destruction of the beta cell and thus delay clinical disease onset. Thus far, attempts have focused on either protecting the beta cell or arresting the immune response, but the future seems to be one of combination therapy. Here, we perform a scoping review on the pathogenesis of T1D, discuss screening strategies, and present promising intervention strategies.
Keywords
TYPE 1 DIABETES: AN AUTOIMMUNE DISEASE WITH A MAJOR ROLE FOR THE BETA CELL ITSELF
Type 1 diabetes (T1D) is an immune-mediated disease in which the insulin-producing beta cells are destroyed by the immune system, resulting in absolute insulin deficiency[1]. The classic hypothesis states that, in a genetically predisposed individual, activation of the immune system by one or multiple environmental triggers results in rapid destruction of the pancreatic beta cells[2]. More recently, attention is shifting towards malfunction of the pancreatic beta cells as a trigger of the immune response, albeit in a genetically at-risk individual[3]. This stresses the importance of genetics in T1D.
With already over 60 loci associated with increased susceptibility to T1D, some genes are linked to beta cell dysfunction and others to immune cell dysfunction, with the HLA region being the major contributor[4,5]. The genetic complexity of T1D is illustrated by the fact that the majority of new T1D diagnoses are made in individuals having no known family history of T1D, despite the 15-fold increased risk for T1D and 2-fold increased risk for coexisting associated autoimmune diseases in individuals having a first-degree relative with T1D[6,7]. In addition, many people carrying the highest risk HLA haplotypes do not develop T1D[8]. However, HLA genes remain the basis of genetic risk prediction models in T1D. The Global Platform for the Prevention of Autoimmune Diabetes (GPPAD) is a European platform that makes use of an enriched genetic risk score (with 47 SNPs) in a general population of newborns to identify those individuals with a 25-fold increased risk of developing T1D (1.3% versus 0.4%, in a Western European background)[9].
The trigger of how T1D occurs in a genetically at-risk individual remains to be elucidated. For years, the immune system was believed to be the only culprit. This issue is from studies in animal models of T1D, such as the NOD mouse that spontaneously develops a disease very similar to T1D, as well as from the detection of autoantibodies against several peptides and proteins of the beta cell in those with recently diagnosed T1D. Moreover, the presence of these autoantibodies in the blood has been shown to have a predictive value for the development of T1D in normoglycemic individuals. These observations were first made in first-degree relatives (and thus genetically predisposed individuals), but they have been extended to the general population. There is now evidence that the presence of two or more autoantibodies almost certainly predicts the evolution to T1D in normoglycemic individuals[10]. Typical autoantibodies are antibodies against glutamic acid decarboxylase (GAD), protein tyrosine phosphatase (IA-2 or ICA512), zinc transporter 8 (ZnT8), and insulin itself[11]. Although these autoantibodies are interesting biomarkers in the prediction of the evolution to T1D, they most likely do not play a pathogenic role. When studying the pancreas of people with T1D who died around the time of diagnosis, it is mainly a cellular infiltrate that is observed (insulitis)[12]. In animal models, the disease is transferred in immune compromised animals by immune cells and not by antibodies[13]. The immune cells responsible for the immune attack remain unknown. However, despite the importance of HLA class II genes in the genetic predisposition, it is not the CD4+ T lymphocyte but rather the HLA class I restricted CD8+ T lymphocyte that is implicated in direct beta cell destruction[14]. However, many other immune cell types could play a role, with an emerging role for innate or non-lymphocyte cells, such as NK cells or neutrophils.
An interesting alternative hypothesis poses that T1D is the result of a dysfunctional beta cell that is cleared by a correctly functioning immune system. In this perspective, T1D etiology becomes comparable to effective anti-tumor immunity and is not classified as an autoimmune disease. Arguments favoring this hypothesis point to a primary defect in insulin-producing beta cells as the initial trigger. This is supported by recent observations suggesting smaller pancreatic volumes in those affected or at-risk of T1D[15,16]. Furthermore, clear signs of beta cell stress can be detected in those on their way to developing T1D, exemplified by an increased proinsulin-to-insulin ratio[17]. This increased ratio suggests abnormalities in insulin processing and vesicular trafficking[18]. Moreover, non-specific triggers associated with T1D, such as increased metabolic demand or viral infections, have been shown to induce endoplasmic reticulum (ER) stress and thereby stress beta cells[19]. This results in a vicious cycle, as ER stress may again increase the visibility of the beta cells to the immune system, thereby initiating further destruction of non-affected islets[20]. Indeed, a beta cell under attack results in the release of additional pro-inflammatory cytokines and chemokines by the beta cells and, as such, attracts even more cells of the immune system[21]. Moreover, beta cells overexpress HLA class I molecules, creating an additional homing beacon for cytotoxic T cells[22]. Whereas both the immune-mediated and the beta cell-centric hypothesis hold their ground, T1D is most likely the result of a complex network of dysfunctions in both the beta cells and the immune system[23]. Interestingly, this is demonstrated, among others, in the observation that stressed beta cells not only misfold insulin but also misprocess other proteins and peptides, leading to the formation of neo-antigens generating novel epitopes[24]. These novel epitopes by themselves then result in an aberrant immune response as they are believed to trigger the peripheral activation of CD4+ and CD8+ autoreactive T lymphocytes[25].
All these different hypotheses [Figure 1] are not only scientifically intriguing but also probably the reason for the limited success of interventions pursuing an arrest of beta cell destruction. Indeed, interventions attempting the restoration of immune tolerance using antigen-specific therapies have failed, and pure immune suppression or modulation has only succeeded in temporarily delaying the decline of functional beta cell mass[26]. These failed interventions also triggered the belief that T1D is a heterogeneous disease, where in some, the immune system may be the causal factor (and then still different immune cell types may be prominent in the attack), whereas in others, more beta cell defects are responsible for the final beta cell disappearance.
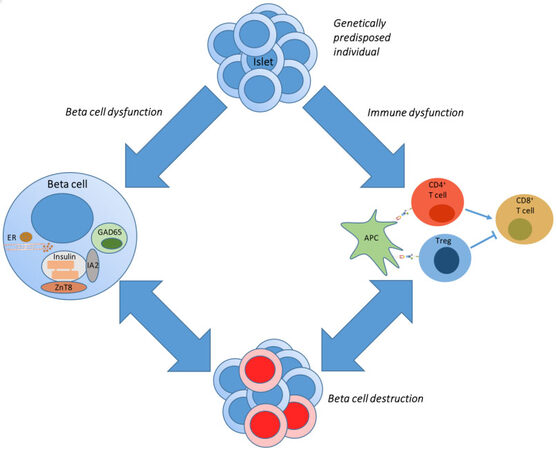
Figure 1. Pathogenesis of type 1 diabetes. In a genetically predisposed individual, a sudden trigger results in beta cell dysfunction or aberrant activation of the immune system, as depicted by the upper blue arrows. This eventually results in the destruction of the beta cells. Beta cell dysfunction can also trigger a (healthy) immune response to clear the dysfunctional beta cells (depicted by double-headed arrows). In a similar way, beta cell destruction by the immune system will cause stress on the remaining beta cells, resulting in further beta cell apoptosis. APC: Antigen-presenting cell; ER: endoplasmic reticulum; GAD: glutamic acid decarboxylase; IA2: islet-antigen 2; Treg: regulatory T cell; ZnT8: zinc-transporter 8.
TYPE 1 DIABETES: SEARCH FOR BIOMARKERS
The presence of islet-specific autoantibodies, not only in genetically predisposed individuals but also in the general population, remains the best predictor of progression towards T1D[27]. Nevertheless, the application of islet-specific autoantibodies as a predictor of T1D is not ready for daily clinical practice. One of the main issues remains the extent of heterogeneity, with the risk of T1D depending on the antibody titer, affinity, immunoglobulin subclasses, and target epitopes on single or multiple islet autoantigens[28]. Insulin autoantibodies (IAA) and GAD autoantibodies (GADA) are most frequently the first appearing autoantibodies, with a higher prevalence of the former in children and the latter in adults[29]. Moreover, the presence of only one islet autoantibody is not sufficient to determine the evolution towards T1D. In a prospective cohort study, only 15% of children with one islet autoantibody developed T1D within 10 years, compared to 70% of those with at least two islet autoantibodies[10]. A recent study showed that autoantibody appearance usually happened before six years of age, with optimal screening ages for initial islet autoantibody screening in children at two and six years of age[30]. Therefore, the consensus states that only the positivity of at least two islet autoantibodies confers a high risk of developing symptomatic T1D[31].
As the above-mentioned tackles the obstacles in the use of islet-specific autoantibodies as a biomarker, the quest for novel biomarkers is on. Novel biomarkers can contribute to a more precise prediction of disease, a better understanding of the heterogeneity of the disease, and predict or at least measure the response to therapy. In INNODIA (Innovative Medicines Initiative of the European Commission; www.innodia.eu), a public–private partnership, over 50 clinical centers in Europe, both pediatric and adult clinics, are collecting samples from new-onset T1D patients and unaffected family members of people living with T1D. In a natural history study spanning several years, modular interrogation platforms for the analysis of cellular and molecular features of beta cell and immune cell biomarkers have been established. These include proteomes, lipidomes, and metabolomes, as well as a full immunome and RNA analyses. A highly standardized collection of samples, followed by highly standardized and quality-controlled analyses in accredited laboratories, should allow robust conclusions when performing an integrated multi-omics natural history study on samples of new-onset T1D individuals or antibody-positive first-degree relatives of people with T1D. Of importance, these biomarker analyses are also included in the clinical trials running in the INNODIA network, thus not only opening the path to biomarkers of disease but also raising hope for the discovery of biomarkers of therapeutic effect and success of interventions.
PREVENTION OR ARREST OF TYPE 1 DIABETES: ARE WE THERE YET?
Why have we not cured T1D yet? It is not because of a lack of trying. The list of studies already performed in people with new-onset T1D or unaffected high-risk family members is already very long. In an attempt to exploit resources and efforts, the scientific community, together with pharmaceutical companies and patient advisors, have joined globally into large consortia. Examples include the Type 1 Diabetes TrialNet [established in 2001 as a National Institutes of Health (NIH)-funded and Juvenile Diabetes Research Foundation (JDRF)-supported international clinical trial network that emerged from the Diabetes Prevention Trial Type 1 (DPT-1)] and the more recent INNODIA consortium (a European partnership among academic institutions, industrial partners, and patient organizations)[32]. These consortia have the advantage of being multi-centered and often have a master protocol, allowing comparison between different studies.
To date, interventions mainly focused on either targeting an ongoing immune response by general or specific immune suppression or modulation [Table 1] or the induction of tolerance to beta cell-related antigens to prevent T1D [Table 2].
Overview of clinical trials targeting arrest of new-onset type 1 diabetes
| Trial | Mechanism | Location | Effect | Phase | DOP/DOFR | Refs. |
| Cyclosporin A | Calcineurin inh | Eu | Transient | 2 | 1986 | [58] |
| Rituximab | Anti-CD20 mAb | Au, Eu, NA | Transient | 2 | 2009 | [59] |
| Mycophenolate + Daclizumab | IMDPH inh + anti-CD25 mAb | NA | No | 2 | 2010 | [60] |
| PROTÉGÉ - Teplizumab | Anti-CD3 mAb | NA | No | 3 | 2011 | [44] |
| Abatacept | CTLA-4-Ig | NA | Transient | 2 | 2011 | [61] |
| AbATE - Teplizumab | Anti-CD3 mAb | NA | Transient | 2 | 2013 | [62] |
| T1DAL - Alefacept | LFA-3-Ig | NA | Potential | 2 | 2013 | [63] |
| DEFEND - Otelixizumab | Anti-CD3 mAb | Eu, NA | No | 3 | 2014 | [42,43] |
| START - High-dose ATG | HD anti-thymocyte globulin | NA | No | 2 | 2016 | [35] |
| Low-dose ATG | LD anti-thymocyte globulin | NA | Beneficial | 2 | 2018 | [37] |
| Verapamil | Calcium channel-blocker | NA | Beneficial | 2 | 2019 | NCT02372253 |
| T1GER - Golimumab | Anti-TNF-α mAb | NA | Beneficial | 2 | 2020 | [50] |
| Anti-IL-21 + Liraglutide | Anti-IL-21 mAb + GLP-1 RA | NA, Eu | Beneficial | 2 | 2021 | [56] |
| Tocilizumab | Anti-IL-6 receptor mAB | Au, NA | Ongoing | 2 | 2021 | NCT02293837 |
| Ladarixin | CXCR1 and CXCR2 Inh | Eu | No | 2 | 2022 | [64] |
| DIABIL-2 - IL-2 | Recombinant human IL-2 | Eu | Ongoing | 2 | / | NCT02411253 |
| ITAD - IL-2 | Recombinant human IL-2 | Eu | Ongoing | 2 | / | NCT03782636 |
| Iscalimab | Anti-CD40 mAb | Eu | Ongoing | 2 | / | NCT04129528 |
| I-DIT - Ixekizumab | Anti-IL-17 mAb | Eu | Ongoing | 2 | / | NCT04589325 |
| UST1D2 - Ustekinumab | Anti-IL-12 / Anti-IL-21 mAb | NA | Ongoing | 2-3 | / | NCT03941132 |
| MELD-ATG | LD anti-thymocyte globulin | Eu | Ongoing | 2 | / | NCT04509791 |
| BANDIT - Baricitinib | JAK1 and JAK2 Inh | Au | Ongoing | 2 | / | NCT04774224 |
Overview of clinical trials using antigen-specific therapy in type 1 diabetes
| Trial | Mechanism | Location | Effect | Phase | DOP/DOFR | Refs. |
| DPT-1 | Parenteral Insulin | NA | No | 3 | 2002 | [65] |
| DPT-1 | Oral Insulin | NA | No | 3 | 2005 | [66] |
| DIPP | Nasal Insulin | Eu | No | 3 | 2008 | [67] |
| Pre-POInT | Oral Insulin | Eu | Potential | 1-2 | 2015 | [68] |
| TrialNet | Oral Insulin | Eu, NA | No | 3 | 2017 | [69] |
| DIAPREV-IT | SC GAD-Alum | Eu | No | 2 | 2018 | [70] |
| Pre-POInT-Early | Oral Insulin | Eu | No | 2 | 2021 | [71] |
| DIAGNODE | ILIT GAD-Alum + Vit D3 | Eu | Potential | 2 | 2021 | [72] |
| DIAPREV-IT 2 | SC GAD-Alum + Vit D3 | Eu | Ongoing | 2 | 2020 | NCT02387164 |
| INIT-II | Nasal Insulin | Au, NZ | Ongoing | 2 | / | NCT00336674 |
| FR1da | Oral Insulin | Eu | Ongoing | 2 | / | NCT02620072 |
| PINIT | Nasal Insulin | Eu | Ongoing | 2 | / | NCT03182322 |
| Oral Proinsulin + IL-10 | Oral Proinsulin + IL-10 | Eu, NA | Ongoing | 1-2 | / | NCT03751007 |
| IMPACT | SC IMCY-0098 | Au, Eu, NA | Ongoing | 2 | / | NCT04524949 |
Interventions arresting an ongoing immune response by general immunosuppression, with cyclosporin A as a prime example, were the first to demonstrate the potential to induce disease remission in new-onset T1D. Despite these promising results, the major obstacles associated with this strategy are disease recurrence and the adverse effects associated with general immunosuppressive drugs (reviewed in[33]). This breakthrough resulted in the quest for an immunosuppressive or immunomodulatory drug that could overcome both obstacles. As such, to date, many immune agents have been tested, with low-dose anti-thymocyte globulin (ATG) and teplizumab (anti-CD3 antibody) being the most promising in people with new-onset T1D[34].
ATG finds its origin in transplantation, and, compared to the relatively higher doses (6.5 mg/kg) used in the randomized controlled Study of Thymoglobulin to ARrest T1D (START) trial, it is found to be more effective in new-onset T1D when used in lower doses (2.5 mg/kg)[35-37]. The protective effect of the lower dose is based on a transient T lymphocyte depletion followed by a T lymphocyte reconstitution in favor of regulatory T lymphocytes, resulting in a shift towards tolerance induction[38]. This is further established by the observation that the addition of G-CSF to a low-dose ATG regimen led to a decrease in the protective effect of ATG[36]. In INNODIA, in phase II, randomized, placebo-controlled, MELD-ATG trial, researchers are testing if even lower doses of ATG would be effective in arresting the decline of functional beta cell mass in people with newly diagnosed T1D (NCT04509791).
In the search for more specific immunomodulatory agents to arrest a T cell-mediated autoimmune disease, attention has shifted to the use of anti-CD3 monoclonal antibodies. The initial clinical pilot trials using humanized anti-CD3 monoclonal antibodies (i.e., teplizumab or the aglycosylated otelixizumab) were hopeful as they showed preservation of beta cell function[39,40]. However, no one could have predicted what followed. Hereafter, large multicenter, randomized, placebo-controlled phase III trials for both otelixizumab and teplizumab failed. Unfortunately, the main reason for this is an alternation of the study protocol in the former and an incorrect choice of the primary endpoint in the latter. Later, this story of anti-CD3 monoclonal antibodies became a prime example of the importance of choosing the correct study protocol and endpoints (reviewed in[41]) and one of the main reasons large consortia often work with a master protocol. For otelixizumab, this was the DEFEND trial, which was probably unsuccessful due to a 15-fold dose reduction in the effective dose[42,43]. For teplizumab, its large phase III trial was the PROTÉGÉ trial. Here, the study population (patients diagnosed with T1D within the past 12 weeks) and the choice of the endpoint (insulin requirement) were the suspected reasons for therapy failure[44]. Later, the randomized, open-label, AbATE trial narrowed the timeframe of new-onset T1D to enroll patients only with a new diagnosis in the past eight weeks and corrected the primary endpoint to a change in C-peptide. In this way, they demonstrated that teplizumab was able to preserve C-peptide in people with new-onset T1D, with a decline in C-peptide up to seven years after diagnosis in responders[45,46].
The AbATE trial demonstrated that an earlier start of therapy resulted in the rescue of more residual beta cells. Based on this, teplizumab was given even earlier in the disease process of T1D, namely in people with stage 2 T1D (defined as the presence of two or more diabetes-related autoantibodies and dysglycemia). Here, teplizumab was able to delay progression to clinical T1D for up to three years[47,48]. These encouraging results have led to the submission of teplizumab to the regulatory authorities for the delay of T1D in prediabetes.
Another lesson learned from the anti-CD3 trials is that an intervention should not be written off too soon. Indeed, for a long time, it was believed that anti-inflammatory interventions targeting single cytokines (e.g., TNF-α or IL-1) are not successful in T1D[49]. However, the recent phase II, multicenter, placebo-controlled, double-blind, T1GER study, where anti-TNF-α receptor antibodies were administered continuously, showed that the decline of functional beta cell mass, as measured by stimulated C-peptide, could be arrested by continued administration of the antibody[50].
Therapies targeting the restoration of tolerance of the immune system towards beta cell-related antigens, such as insulin or GAD, have failed to be successful[51]. However, this approach remains interesting, as it holds the potential to induce longer-lasting beta cell protection if one could restore tolerance to the beta cell. Hypothesizing that a combination of immune modulation (“clearing the autoimmune attack at the time of T1D diagnosis”) and antigen administration (“restoring tolerance”) could offer the perspective of long-term effect, several approaches have been designed, and some tested. One of these is the administration of proinsulin (as an autoantigen) in combination with the cytokine IL-10 through a genetically engineered Lactococcus lactis as the carrier. This approach allows administering this antigen via an oral route (a pathway known for tolerance induction), and interim results of the phase Ib (open-label) and IIa (randomized, double-blind), multicenter, study with this approach (AG019 ActobioticsTM) demonstrate both safety and potentially interesting immune effects[52] (NCT03751007). The investigators even brought in an additional immune modulator in the form of teplizumab, based on successful animal studies[53,54], which again showed good safety and interesting immune effects. Another promising combination of immune modulation and antigen was tested in the DIAGNODE-1 pilot trial, where GAD dissolved in alum was administered into lymph nodes (a more targeted approach), as well as in combination with vitamin D for low-grade immunomodulation[55]. Based on relatively promising results, GAD in alum is now being tested in combination with ibuprofen (DIABGAD, an interventional pilot trial; NCT01785108), etanercept (EDCR, an interventional open-label trial; NCT02464033), or GABA (GABA/Diamyd, a randomized placebo-controlled trial; NCT02002130) as an anti-inflammatory component.
Finally, based on the observations described above, suggesting a role in the pathogenesis of T1D for both the beta cells and the immune system, other trials have been conducted in an attempt to combine immune modulation with beta cell protective agents. A recent randomized, placebo-controlled, phase II trial combined liraglutide, a GLP-1 receptor agonist, and an antibody targeting the cytokine IL-21. This combination therapy showed a clear delay in the decline of functional beta cell mass[56].
FUTURE PERSPECTIVES
Why have we not succeeded in arresting T1D? In the most popular animal model, the NOD mouse, the list of successful interventions is long, but a careful reading of the literature shows that these interventions are particularly successful when administered early in the life of the mouse before any autoimmune attack has started[57]. In humans, however, this would be the equivalent of treating newborns. As long as our predictive power is low (genetic risk scores allow enrichment in the general population of up to 25-fold, but they still only provide a risk attribution of around 1%), only very safe interventions will be tolerated. As such, on the GPPAD platform, the cross-sectional cohort Freder1k trial is evaluating the effect of administration of oral insulin on the onset of autoimmunity (autoantibodies against the beta cell) (NCT03316261), and the multicenter, randomized, placebo-controlled, SINT1A study is evaluating the impact of probiotics on islet autoimmunity development (NCT04769037). A major issue with these early interventions is the number of people to be screened, as well as the amount of time these people need to be followed up. Even when individuals at later stages of T1D (such as those with autoantibodies with or without dysglycemia stage 1/2), and thus higher risk to progress towards clinical T1D (stage 3), are studied, these trials are logistically challenging due to the large cohorts and long follow-up needed. In the future, the combined use of immune interventions together with beta cell augmenting and/or replacement therapy, such as stem cell-based therapy, might offer a solution to compensate for the lost beta cell function at the time of clinical diagnosis.
CONCLUSIONS
Type 1 diabetes remains one of the most common and severe chronic diseases in children, adolescents, and young adults. However, despite its high prevalence, we keep on using a standard therapy that is already more than a century old. Technological advances have helped make giant strides in regulating optimal glucose homeostasis, but the chronic burden of long-term hyperglycemia-related complications remains troublesome. Despite all efforts, the road to a cure for T1D remains long and full of obstacles. The first is the need for biomarkers allowing early screening. The second is the need for superior treatment options, as this pursuit for early biomarkers should not turn into a Sword of Damocles. Whereas interventions thus far have mainly focused on either targeting the ongoing immune response or the induction of tolerance to the beta cell, we believe future research should focus on targeting both. A limitation to this review is that the field of T1D research is extensive and ever ongoing (as the long list of currently ongoing trials shows). Nevertheless, we hope this review shows that the pursuit of early biomarkers can go hand in hand with superior treatment options, as early screening can result in an earlier treatment initiation at a time of a higher residual functional beta cell mass. Thus, when asking the question, “Are we there yet?”, we have to say, “Not yet”. However, the future does look bright as we have high hopes for the trend towards global consortia as joined effort and pooled resources hopefully have a synergistic effect.
DECLARATIONS
Authors’ contributionsConceptualized the review goals and wrote the manuscript: Mathieu C, Martens PJ
All authors contributed to the article, consent to participate and approved the submitted version.
Availability of data and materialsThe data that support the findings of this study are openly available in PubMed at https://pubmed.ncbi.nlm.nih.gov/.
Financial support and sponsorshipThis work was supported by IMI2-JU under grant agreements 115797 (INNODIA) and 945268 (INNODIA HARVEST). This joint undertaking receives support from the European Union’s Horizon 2020 research and innovation program and European Federation of Pharmaceutical Industries and Associations, JDRF, and The Leona M. and Harry B. Helmsley Charitable Trust and KU Leuven (C16/18/006). This publication was supported by the Open Access Publication Fund of the KU Leuven.
Conflicts of interestThe authors declared that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022
REFERENCES
1. Atkinson MA, Eisenbarth GS. Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet 2001;358:221-9.
2. Eisenbarth GS. Type I diabetes mellitus. A chronic autoimmune disease. N Engl J Med 1986;314:1360-8.
3. Atkinson MA, Bluestone JA, Eisenbarth GS, et al. How does type 1 diabetes develop?: the notion of homicide or β-cell suicide revisited. Diabetes 2011;60:1370-9.
4. Bakay M, Pandey R, Grant SFA, Hakonarson H. The genetic contribution to type 1 diabetes. Curr Diab Rep 2019;19:116.
5. Santin I, Eizirik DL. Candidate genes for type 1 diabetes modulate pancreatic islet inflammation and β-cell apoptosis. Diabetes Obes Metab 2013;15 Suppl 3:71-81.
7. Milluzzo A, Falorni A, Brozzetti A, et al. Risk for coexistent autoimmune diseases in familial and sporadic type 1 diabetes is related to age at diabetes onset. Endocr Pract 2021;27:110-7.
8. Skyler JS, Bakris GL, Bonifacio E, et al. Differentiation of diabetes by pathophysiology, natural history, and prognosis. Diabetes 2017;66:241-55.
9. Winkler C, Haupt F, Heigermoser M, et al. GPPAD Study Group. Identification of infants with increased type 1 diabetes genetic risk for enrollment into Primary Prevention Trials-GPPAD-02 study design and first results. Pediatr Diabetes 2019;20:720-7.
10. Ziegler AG, Rewers M, Simell O, et al. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA 2013;309:2473-9.
12. Pugliese A. Insulitis in the pathogenesis of type 1 diabetes. Pediatr Diabetes 2016;17 Suppl 22:31-6.
13. De Leenheer E, Wong FS. Adoptive transfer of autoimmune diabetes using immunodeficient Nonobese Diabetic (NOD) mice. In: Gillespie KM, editor. Type-1 Diabetes. New York: Springer; 2016. pp. 135-40.
14. Burrack AL, Martinov T, Fife BT. T cell-mediated beta cell destruction: autoimmunity and alloimmunity in the context of type 1 diabetes. Front Endocrinol (Lausanne) 2017;8:343.
15. Virostko J, Williams J, Hilmes M, et al. Pancreas volume declines during the first year after diagnosis of type 1 diabetes and exhibits altered diffusion at disease onset. Diabetes Care 2019;42:248-57.
16. Campbell-Thompson ML, Filipp SL, Grajo JR, et al. Relative pancreas volume is reduced in first-degree relatives of patients with type 1 diabetes. Diabetes Care 2019;42:281-7.
17. Wasserfall C, Nick HS, Campbell-Thompson M, et al. Persistence of pancreatic insulin mRNA expression and proinsulin protein in type 1 diabetes pancreata. Cell Metab 2017;26:568-575.e3.
18. Rodriguez-Calvo T, Zapardiel-Gonzalo J, Amirian N, et al. Increase in pancreatic proinsulin and preservation of β-cell mass in autoantibody-positive donors prior to type 1 diabetes onset. Diabetes 2017;66:1334-45.
19. Eizirik DL, Miani M, Cardozo AK. Signalling danger: endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. Diabetologia 2013;56:234-41.
20. Thomaidou S, Kracht MJL, van der Slik A, et al. β-cell stress shapes CTL immune recognition of preproinsulin signal peptide by posttranscriptional regulation of endoplasmic reticulum aminopeptidase 1. Diabetes 2020;69:670-80.
21. Cardozo AK, Proost P, Gysemans C, Chen MC, Mathieu C, Eizirik DL. IL-1beta and IFN-gamma induce the expression of diverse chemokines and IL-15 in human and rat pancreatic islet cells, and in islets from pre-diabetic NOD mice. Diabetologia 2003;46:255-66.
22. Richardson SJ, Rodriguez-Calvo T, Gerling IC, et al. Islet cell hyperexpression of HLA class I antigens: a defining feature in type 1 diabetes. Diabetologia 2016;59:2448-58.
23. Peters L, Posgai A, Brusko TM. Islet-immune interactions in type 1 diabetes: the nexus of beta cell destruction. Clin Exp Immunol 2019;198:326-40.
24. Kracht MJL, Zaldumbide A, Roep BO. Neoantigens and microenvironment in type 1 diabetes: lessons from antitumor immunity. Trends Endocrinol Metab 2016;27:353-62.
25. Roep BO, Kracht MJ, van Lummel M, Zaldumbide A. A roadmap of the generation of neoantigens as targets of the immune system in type 1 diabetes. Curr Opin Immunol 2016;43:67-73.
26. Martens PJ, Gysemans C, Mathieu C. 100 YEARS OF INSULIN: arresting or curing type 1 diabetes: an elusive goal, but closing the gap. J Endocrinol 2021;249:T1-T11.
27. Primavera M, Giannini C, Chiarelli F. Prediction and prevention of type 1 diabetes. Front Endocrinol (Lausanne) 2020;11:248.
28. Bonifacio E, Achenbach P. Birth and coming of age of islet autoantibodies. Clin Exp Immunol 2019;198:294-305.
29. Vehik K, Bonifacio E, Lernmark Å, et al. TEDDY Study Group. Hierarchical order of distinct autoantibody spreading and progression to type 1 diabetes in the TEDDY study. Diabetes Care 2020;43:2066-73.
30. Ghalwash M, Dunne JL, Lundgren M, et al. Two-age islet-autoantibody screening for childhood type 1 diabetes: a prospective cohort study. Lancet Diabetes Endocrinol 2022;10:589-96.
31. Insel RA, Dunne JL, Atkinson MA, et al. Staging presymptomatic type 1 diabetes: a scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 2015;38:1964-74.
33. Flores C, Fouquet G, Moura IC, Maciel TT, Hermine O. Lessons to learn from low-dose cyclosporin-A: a new approach for unexpected clinical applications. Front Immunol 2019;10:588.
34. Jacobsen LM, Bundy BN, Greco MN, et al. Comparing beta cell preservation across clinical trials in recent-onset type 1 diabetes. Diabetes Technol Ther 2020;22:948-53.
35. Gitelman SE, Gottlieb PA, Felner EI, et al. ITN START Study Team. Antithymocyte globulin therapy for patients with recent-onset type 1 diabetes: 2 year results of a randomised trial. Diabetologia 2016;59:1153-61.
36. Haller MJ, Long SA, Blanchfield JL, et al. Type 1 Diabetes TrialNet ATG-GCSF Study Group. Low-dose anti-thymocyte globulin preserves C-peptide, reduces HbA1c, and increases regulatory to conventional T-cell ratios in new-onset type 1 diabetes: two-year clinical trial data. Diabetes 2019;68:1267-76.
37. Haller MJ, Schatz DA, Skyler JS, et al. Type 1 Diabetes TrialNet ATG-GCSF Study Group. Low-dose Anti-Thymocyte Globulin (ATG) preserves β-cell function and improves HbA1c in new-onset type 1 diabetes. Diabetes Care 2018;41:1917-25.
38. Lu Y, Suzuki J, Guillioli M, Umland O, Chen Z. Induction of self-antigen-specific Foxp3+ regulatory T cells in the periphery by lymphodepletion treatment with anti-mouse thymocyte globulin in mice. Immunology 2011;134:50-9.
39. Herold KC, Hagopian W, Auger JA, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med 2002;346:1692-8.
40. Keymeulen B, Vandemeulebroucke E, Ziegler AG, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med 2005;352:2598-608.
41. Chatenoud L. A future for CD3 antibodies in immunotherapy of type 1 diabetes. Diabetologia 2019;62:578-81.
42. Aronson R, Gottlieb PA, Christiansen JS, et al. DEFEND Investigator Group. Low-dose otelixizumab anti-CD3 monoclonal antibody DEFEND-1 study: results of the randomized phase III study in recent-onset human type 1 diabetes. Diabetes Care 2014;37:2746-54.
43. Ambery P, Donner TW, Biswas N, Donaldson J, Parkin J, Dayan CM. Efficacy and safety of low-dose otelixizumab anti-CD3 monoclonal antibody in preserving C-peptide secretion in adolescent type 1 diabetes: DEFEND-2, a randomized, placebo-controlled, double-blind, multi-centre study. Diabet Med 2014;31:399-402.
44. Sherry N, Hagopian W, Ludvigsson J, et al. Teplizumab for treatment of type 1 diabetes (Protégé study): 1-year results from a randomised, placebo-controlled trial. Lancet 2011;378:487-97.
45. Hagopian W, Ferry RJ Jr, Sherry N, et al. Protégé Trial Investigators. Teplizumab preserves C-peptide in recent-onset type 1 diabetes: two-year results from the randomized, placebo-controlled Protégé trial. Diabetes 2013;62:3901-8.
46. Perdigoto AL, Preston-Hurlburt P, Clark P, et al. Immune Tolerance Network. Treatment of type 1 diabetes with teplizumab: clinical and immunological follow-up after 7 years from diagnosis. Diabetologia 2019;62:655-64.
47. Herold KC, Bundy BN, Long SA, et al. Type 1 Diabetes TrialNet Study Group. An anti-CD3 antibody, teplizumab, in relatives at risk for type 1 diabetes. N Engl J Med 2019;381:603-13.
48. Sims EK, Bundy BN, Stier K, et al. Type 1 Diabetes TrialNet Study Group. Teplizumab improves and stabilizes beta cell function in antibody-positive high-risk individuals. Sci Transl Med 2021;13:eabc8980.
49. Donath MY, Dinarello CA, Mandrup-Poulsen T. Targeting innate immune mediators in type 1 and type 2 diabetes. Nat Rev Immunol 2019;19:734-46.
50. Quattrin T, Haller MJ, Steck AK, et al. T1GER Study Investigators. Golimumab and beta-cell function in youth with new-onset type 1 diabetes. N Engl J Med 2020;383:2007-17.
51. Ludvigsson J. Autoantigen treatment in type 1 diabetes: unsolved questions on how to select autoantigen and administration route. Int J Mol Sci 2020;21:1598.
52. Mathieu C. AG019 ActoBiotics as monotherapy or in association with teplizumab in recent-onset type 1 diabetes was safe and demonstrated encouraging metabolic and immunological effects. Abstract at EASD 57th Annual meeting. 2021. Available from: https://investors.precigen.com/events/event-details/european-association-study-diabetes-easd-57th-annual-meeting/ [Last accessed on 15 Sep 2022].
53. Takiishi T, Cook DP, Korf H, et al. Reversal of diabetes in NOD mice by clinical-grade proinsulin and IL-10-secreting lactococcus lactis in combination with low-dose anti-CD3 depends on the induction of Foxp3-positive T cells. Diabetes 2017;66:448-59.
54. Cook DP, Cunha JPMCM, Martens PJ, et al. Intestinal delivery of proinsulin and IL-10 via lactococcus lactis combined with low-dose anti-CD3 restores tolerance outside the window of acute type 1 diabetes diagnosis. Front Immunol 2020;11:1103.
55. Tavira B, Barcenilla H, Wahlberg J, Achenbach P, Ludvigsson J, Casas R. Intralymphatic glutamic acid decarboxylase-alum administration induced Th2-like-specific immunomodulation in responder patients: a pilot clinical trial in type 1 diabetes. J Diabetes Res 2018;2018:9391845.
56. von Herrath M, Bain SC, Bode B, et al. Anti-interleukin-21 antibody and liraglutide for the preservation of β-cell function in adults with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Diabetes Endocrinol 2021;9:212-24.
57. Shoda LK, Young DL, Ramanujan S, et al. A comprehensive review of interventions in the NOD mouse and implications for translation. Immunity 2005;23:115-26.
58. Feutren G, Assan R, Karsenty G, et al. Cyclosporin increases the rate and length of remissions in insulin-dependent diabetes of recent onset. Lancet 1986;328:119-24.
59. Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, et al. Type 1 Diabetes TrialNet Anti-CD20 Study Group. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med 2009;361:2143-52.
60. Gottlieb PA, Quinlan S, Krause-Steinrauf H, et al. Type 1 Diabetes TrialNet MMF/DZB Study Group. Failure to preserve beta-cell function with mycophenolate mofetil and daclizumab combined therapy in patients with new- onset type 1 diabetes. Diabetes Care 2010;33:826-32.
61. Orban T, Bundy B, Becker DJ, et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet 2011;378:412-9.
62. Herold KC, Gitelman SE, Ehlers MR, et al. AbATE Study Team. Teplizumab (anti-CD3 mAb) treatment preserves C-peptide responses in patients with new-onset type 1 diabetes in a randomized controlled trial: metabolic and immunologic features at baseline identify a subgroup of responders. Diabetes 2013;62:3766-74.
63. Rigby MR, Dimeglio LA, Rendell MS, et al. Targeting of memory T cells with alefacept in new-onset type 1 diabetes (T1DAL study): 12 month results of a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Diabetes Endocrinol 2013;1:284-94.
64. Piemonti L, Keymeulen B, Gillard P, et al. Ladarixin, an inhibitor of the interleukin-8 receptors CXCR1 and CXCR2, in new-onset type 1 diabetes: a multicentre, randomized, double-blind, placebo-controlled trial. Diabetes Obes Metab 2022;24:1840-9.
65. Prevention Trial--Type 1 Diabetes Study Group. Effects of insulin in relatives of patients with type 1 diabetes mellitus. N Engl J Med 2002;346:1685-91.
66. Skyler JS, Krischer JP, Wolfsdorf J, et al. Effects of oral insulin in relatives of patients with type 1 diabetes: the diabetes prevention trial--type 1. Diabetes Care 2005;28:1068-76.
67. Näntö-salonen K, Kupila A, Simell S, et al. Nasal insulin to prevent type 1 diabetes in children with HLA genotypes and autoantibodies conferring increased risk of disease: a double-blind, randomised controlled trial. Lancet 2008;372:1746-55.
68. Bonifacio E, Ziegler AG, Klingensmith G, et al. Pre-POINT Study Group. Effects of high-dose oral insulin on immune responses in children at high risk for type 1 diabetes: the Pre-POINT randomized clinical trial. JAMA 2015;313:1541-9.
69. Krischer JP, Schatz DA, Bundy B, Skyler JS, Greenbaum CJ. Writing Committee for the Type 1 Diabetes TrialNet Oral Insulin Study Group. Effect of oral insulin on prevention of diabetes in relatives of patients with type 1 diabetes: a randomized clinical trial. JAMA 2017;318:1891-902.
70. Larsson H, Lundgren M, Jonsdottir B, Cuthbertson D, Krischer J; DiAPREV-IT Study Group. Safety and efficacy of autoantigen-specific therapy with 2 doses of alum-formulated glutamate decarboxylase in children with multiple islet autoantibodies and risk for type 1 diabetes: a randomized clinical trial. Pediatr Diabetes 2018;19:410-9.
71. Assfalg R, Knoop J, Hoffman KL, et al. Oral insulin immunotherapy in children at risk for type 1 diabetes in a randomised controlled trial. Diabetologia 2021;64:1079-92.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Mathieu C, Martens PJ. Arresting type 1 diabetes: are we there yet? Obstacles and opportunities. Metab Target Organ Damage 2022;2:15. http://dx.doi.org/10.20517/mtod.2022.16
AMA Style
Mathieu C, Martens PJ. Arresting type 1 diabetes: are we there yet? Obstacles and opportunities. Metabolism and Target Organ Damage. 2022; 2(4): 15. http://dx.doi.org/10.20517/mtod.2022.16
Chicago/Turabian Style
Mathieu, Chantal, Pieter-Jan Martens. 2022. "Arresting type 1 diabetes: are we there yet? Obstacles and opportunities" Metabolism and Target Organ Damage. 2, no.4: 15. http://dx.doi.org/10.20517/mtod.2022.16
ACS Style
Mathieu, C.; Martens P.J. Arresting type 1 diabetes: are we there yet? Obstacles and opportunities. Metab Target Organ Damage. 2022, 2, 15. http://dx.doi.org/10.20517/mtod.2022.16
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 18 clicks
Cite This Article 18 clicks

 Like This Article 4
likes
Like This Article 4
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.