
Mechanobiology in the development and progression of nonalcoholic fatty liver disease: an updated review

Abstract
Mechanobiology is a rapidly emerging field focused on the biological impact of physical forces at the molecular, cellular, and tissue level. Living cells perceive mechanical cues and transform them into biochemical signals through mechanotransduction. Mechanotransduction is a complex process that involves mechanosensors (which are located in the plasma membrane or within the cell) and mechanotransmission to the nucleus (which occurs either by physical connection between the mechanosensor and the nucleus or by mechanosignaling through biochemical pathways). Essential biological functions, including development, growth, motility, and metabolism, depend on the mechanoresponses generated by these events. Multiple lines of evidence indicate that disruption of mechanical homeostasis may contribute to the pathogenesis of nonalcoholic fatty liver disease (NAFLD), a highly prevalent metabolic disorder characterized by abnormal accumulation of lipid droplets in hepatocytes (steatosis) and often associated with inflammation and liver cell injury (steatohepatitis). While predicting individual predisposition to adverse outcomes in NAFLD remains a challenge, there is increasing evidence that steatosis and steatohepatitis trigger mechanoresponses that contribute to the early stages of pathogenesis in NAFLD and critically impact disease progression. Lipid accumulation and lipotoxicity modify liver viscoelasticity, alter the biomechanics of liver sinusoids, and initiate aberrant pathways of mechanotransduction in hepatocytes and non-parenchymal liver cells, such as sinusoidal endothelial cells and hepatic stellate cells. Interactions of these cells at mechanical interfaces with each other, with extracellular matrix, and with sinusoidal blood flow are profoundly altered by steatosis and steatohepatitis; such changes may promote a pro-angiogenic and pro-fibrotic milieu. A better understanding of liver mechanobiology may facilitate the identification of novel molecular and cellular targets in the management of NAFLD.
Highlights
● Cellular and molecular behavior is regulated by a variety of physical forces;
● Viscoelastic properties of the liver are altered in nonalcoholic fatty liver disease (NAFLD);
● Sinusoidal hemostasis is disrupted by early functional and structural changes in NAFLD;
● Mechanical cues are likely to contribute to all aspects of NAFLD pathogenesis.
Keywords
INTRODUCTION
The interdisciplinary field of mechanobiology aims to understand how physical forces change the mechanical properties of living organisms and contribute to the regulation of biological functions including development, metabolism, growth, and proliferation[1]. While the role of mechanical factors in the pathobiology of hard tissues (such as bone or cartilage) is well established, increasing evidence suggests that mechanical cues drive many cellular and molecular mechanisms within soft tissues such as the liver[2,3]. Indeed, all living cells have the ability to sense and generate physical forces while interacting with their microenvironment[4]. This dialog can be described within the context of mechanical homeostasis, which is essential for maintaining normal cell functions and regulating cell fate[5]. The process in which physical forces arising both inside and outside of the cell evoke biochemical and transcriptional responses is termed mechanotransduction[6,7]. Mechanotransduction is a complex process that involves mechanosensors (which are located in the plasma membrane or within the cell) and mechanotransmission to the nucleus (which may be mediated either by physical linkage between the mechanosensor and the nucleus via the contractile cytoskeleton or by mechanosignaling through biochemical pathways, including enzymatic cascades and soluble mediators)[1,6,8,9].
nonalcoholic fatty liver disease (NAFLD) is a metabolic disorder rooted in caloric excess and initiated by steatosis with the potential to culminate in end-stage liver disease or liver cancer[10]. From the cellular to the organ level, the physical properties of the liver are altered in NAFLD[3,11,12]. Accumulation of lipid droplets disrupts normal biomechanics of the individual hepatocytes, the liver microcirculation, Clearly, liver is an organ, and the liver at the organ-level.[13-15]. Moreover, steatosis is often associated with pathologic events that further perturb the mechanical microenvironment in the liver, including inflammation, endothelial dysfunction, impaired vasoregulation, neoangiogenesis, and fibrosis[16,17]. While mechanotransduction as a key process associated with liver fibrosis has received increasing attention[18-21], the contribution of steatosis-related mechanical changes to the progression of NAFLD remains relatively unexplored. Here, we review recent advances in our understanding of the interplay between liver biomechanics and the pathobiology of NAFLD with a focus on the early stages of disease development.
PRINCIPLES AND COMPONENTS OF VISCOELASTICITY IN THE LIVER
In general physical terms, objects exposed to physical forces respond with various degrees of deformation. In solid materials, this phenomenon is described by the stress-strain relationship, in which stress σ denotes the magnitude of force F per unit area A (σ = F/A) and strain ε denotes the fraction of change ΔL per unit length L as a dimensionless quantity (ε = ΔL/L). Elasticity or stiffness is a key attribute of solid materials, indicating their ability to resist deformation in response to physical forces[22]. Elasticity also refers to the ability of an object to return to its original shape and size when the forces causing its deformation are removed. The stress-strain relationship in solid materials is linear at low stress levels and can be characterized by the E elastic modulus or Young’s modulus (E = σ/ε)[3]. As stress continues to increase, solid materials may reach their fracture point and break. In contrast, the deformation of fluids is characterized by viscosity, which is defined as the resistance of liquids or gases to flow or spread. In considering a fluid phase between two boundary plates (solid phases), the force per unit area required to move one plate over the other at a velocity u and at a distance y from the stationary plate will depend on the dynamic viscosity μ, defined by the internal shear stress or friction between adjacent fluid layers according to the formula
Most living cells and tissues can be described as viscoelastic, indicating that their physical properties are determined by coexisting solid and fluid components[3]. Accordingly, soft tissues such as the liver are not purely elastic or purely viscous and show inherently more complex (non-linear) mechanical and rheological characteristics[23]. The complex modulus of viscoelasticity (G*) is defined as a combination of the storage modulus (G’, reflecting stiffness) and the loss modulus (G”, reflecting viscosity)[24]. Stress relaxation (viscous dissipation) is an attribute of viscoelastic materials that describes how stress may gradually decrease with time due to improved ability of the system to withstand deformation before reaching the fracture point. In other words, relaxation denotes a time-dependent reduction in stress under a constant level of strain[25]. As a counterpart, creep compliance describes the extent of time-dependent deformation (“cold flow”) of viscoelastic materials as the rate at which strain increases upon the application of constant stress[22].
From a simplified mechanobiological perspective, the liver is a highly vascular organ with three major compartments: (i) intravascular (blood, lymph) and transcellular (bile) fluids; (ii) liver cells including hepatocytes and non-parenchymal cells; and (iii) extracellular matrix (ECM) including interstitial fluid. These components are organized into complex structural and functional units in which various liver cells receive and process diverse mechanical information. The mechanical properties, relative proportion, and interactions of these tissue constituents are important determinants of liver structure and function in health and disease[24,26].
At any given time, the liver contains 10%-15% of the total blood volume; within the liver, 60% of this blood volume is found in the specialized liver capillaries called sinusoids[27]. Liver sinusoids are low-flow, low-pressure vascular channels surrounded by the cellular and ECM compartments of the liver[27]. The hepatic artery supplies one-third of hepatic blood flow, while the portal vein, which collects nutrient-rich blood from the splanchnic vessels of the gastrointestinal tract, supplies the remaining two-thirds. Terminal branches of the hepatic artery and the portal vein merge into the sinusoids, which extend from the portal tract to the central vein.
As the main cell type of the liver, hepatocytes make up 80% of the liver mass and are organized into interconnected plates, which form hexagonal lobules around the central vein. Liver sinusoidal endothelial cells (LSECs) form the wall of sinusoidal vascular channels. The basement membrane of LSECs is fenestrated and lacks basal lamina, making these cells highly permeable[28]. Sinusoidal fenestrae cluster into sieve plates that are closely associated with the underlying actin cytoskeleton of LSECs[29]. LSECs define the space of Disse, which contains ECM and separates hepatocytes from LSECs and sinusoidal blood [Figure 1]. Hepatic stellate cells (HSCs) are located in the space of Disse and have long cellular projections that encircle one or more sinusoids similar to pericytes in the systemic circulation[30]. Kupffer cells are liver macrophages that reside in the sinusoidal lumen and are especially prominent in the vicinity of the portal confluence where they recognize molecular danger signals derived from the portal circulation[31].
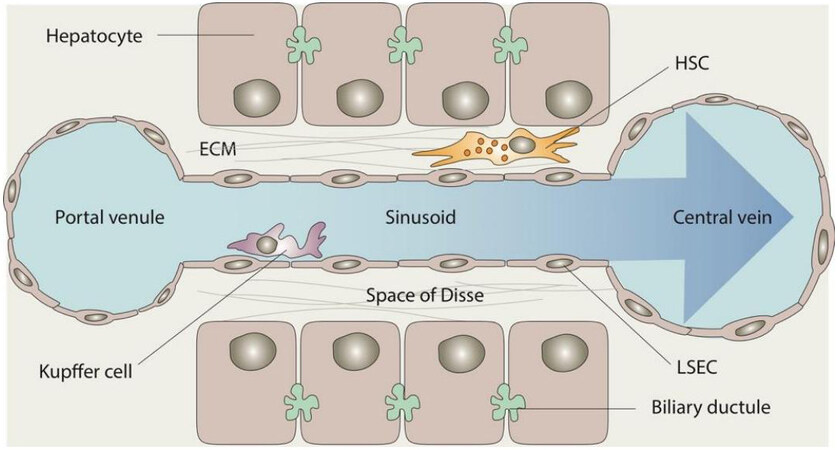
Figure 1. Structure of the liver sinusoid with key cellular components. ECM: Extracellular matrix; HSC: hepatic stellate cell; LSEC: liver sinusoidal endothelial cell.
The extracellular matrix (ECM) is essential to hepatic architecture and physiology[26]. ECM is an insoluble, noncellular complex composed of water, proteoglycans, glycoproteins, and fibrillar proteins that provide a physical scaffold in multicellular organisms[32]. Dynamic components of the viscoelastic matrix interact with mechanosensitive cell surface receptors (e.g., focal adhesions) and influence cell differentiation, proliferation, migration, and metabolism[6,33]. In a healthy liver, ECM forms the dense connective tissue layer of Glisson’s capsule surrounding the liver and contributes to the boundary between sinusoidal blood and hepatocytes in the space of Disse, but is otherwise limited[34]. With the gradual expansion and fibrotic transformation of ECM, elasticity, rather than viscosity, increasingly dominates the matrix mechanical properties, which in turn, affects cell behavior[35].
The coexistence of viscosity and elasticity in the liver tissue represents a diagnostic challenge to the evaluation of NAFLD. A variety of ultrasound-based methods, such as vibration-controlled transient elastography (VCTE) and shear wave elastography (SWE), have been developed to reduce the need for liver biopsy by non-invasively assessing and monitoring chronic liver disease[36]. The primary goal of these diagnostic tools is to determine the degree of fibrosis, which closely correlates with shear wave velocity and is a key predictor of progression in NAFLD[37]. However, additional shear wave characteristics may provide useful information about other aspects of NAFLD, including pre-fibrotic NAFLD. It has been shown that shear wave attenuation (SWA) increases with the degree of steatosis[38], while shear wave dispersion (SWD) predicts the degree of necro-inflammation[39]. In fact, detection of the controlled attenuation parameter (CAP) based on these principles has become a standard method for the noninvasive assessment of hepatic fat content[40]. However, separated extraction of the elastic and viscous components of the liver from the shear wave analysis remains challenging[41], and improvement of these methods is an active area of research[42,43]. A more detailed discussion of this rapidly evolving field is beyond the scope of this review.
MECHANOBIOLOGY OF LIPID DROPLET ACCUMULATION IN LIVER CELLS
Lipid droplets (LDs) are dynamic organelles consisting of an inner core of neutral lipids (predominately triacylglycerols and esterified cholesterol) surrounded by a phospholipid monolayer and primarily functioning as energy depots in the cell[13]. As adipocytes mature, they substantially enlarge in size (hypertrophy) due to the significant accumulation of LDs, which continue to grow and often merge, inflating the cell and stretching the plasma membrane[44]. These events significantly change the viscoelastic characteristics of mature adipocytes. Studies using atomic force microscopy (AFM), which is a nanoscale tool for the analysis of biomechanical characteristics at the single-cell level, have found that LDs are 2.5 to 8.3 folds stiffer than the cytoplasm, indicating that higher proportions of LDs lead to increased cellular stiffness and distortion of the intracellular environment in adipocytes[45].
While accumulation of LDs is a physiological process during the maturation of adipocytes, the presence of LDs exceeding 5% of the cross-sectional area of the liver is considered pathological[46]. Hepatocellular enlargement due to lipid accumulation may reduce sinusoidal space by as much as 50% compared to a healthy liver[47]. Indeed, in severe steatosis, lipid-laden and engorged hepatocytes compress on the sinusoidal space, leading to tortuous and narrowed sinusoids[48,49]. Deficiencies of various enzymes and structural proteins involved in the formation, expansion, and degradation of LDs have been implicated in hepatocellular fat accumulation, which may manifest as macrovesicular steatosis (characterized by one or more large LDs displacing the nucleus) or microvesicular steatosis (characterized by the presence of numerous small LDs that do not displace the nucleus)[50]. Genetic variants associated with increased risk of NAFLD have been identified for perilipin 2 (PLIN2), which helps to stabilize LDs and inhibit autophagy of growing LDs[51]; for the patatin-like phospholipase domain-containing protein 3 (PNPLA3), which helps to remove triacylglycerols from the LD core and facilitate the proteasomal degradation of LDs[52]; and for the protein product of transmembrane 6 family member 2 (TM6SF2), which is normally involved in the secretion of triacylglycerol-rich lipoproteins[53]. The biology of LDs and their involvement in the pathophysiology of NAFLD have been summarized in several recent and excellent reviews[13-15]. Here we focus on the biomechanical aspects of LDs relevant to steatosis and steatohepatitis.
Multiple efforts have been made to better understand the effects of microvesicular and macrovesicular steatosis on viscoelasticity at the single-cell level. As in the adipocyte studies referenced above, AFM has been used to characterize the mechanical properties of single liver cells (human hepatoma cell line HepG2) containing different amounts of fat. In one study, liver cells were exposed to increasing concentrations of oleic acid to produce dose-dependent changes in intracellular lipid deposition[54]. In cells exposed to higher oleic acid concentrations, LDs were noted to fuse into larger LDs and distort the nucleus. The single-cell stress-strain relationship was defined through creep compliance, which positively correlated with increasing lipid concentrations and yielded predictable changes in viscoelasticity of the single cell, even when lipid content was low[54]. Interestingly, staining of F-actin in fat-loaded HepG2 cells indicated no discernible changes in relation to lipid deposition, suggesting that the observed viscoelastic changes were not due to cytoskeletal remodeling[54]. The authors concluded that liver cells tend to be fluid-like when their lipid content increases, with less stiffness and more viscosity. While these findings are at odds with earlier studies on adipocyte stiffness[45], it is unclear whether the differences relate to cell type, experimental methodology, or detection differences. Given that the single-cell AFM method removes the complexity of the cell’s mechanical microenvironment, using single-cell level changes to make predictions about the mechanical properties of the organ as a whole is challenging.
AFM and single-cell force spectroscopy has also been used to investigate how steatosis of different types and severity affects cellular biomechanics[55]. In one study, rat FaO hepatoma cells were exposed to different steatosis-triggering agents, including fatty acids, fructose, a combination of fatty acids and fructose, or a combination of fatty acids and tumor necrosis factor-alpha (TNFα). The largest increase in the size of LDs (334% as compared to controls) was seen with exposure to the combination of fructose and fatty acids, while the largest increase in the number of LDs per cell (9-fold as compared to controls), along with the largest increase in markers of ER stress and hepatocyte damage, was seen with exposure to the combination of fatty acids and TNFα. The relative elasticity (Er) of single cells, as assessed by single-cell force spectroscopy, was significantly increased in all exposure groups compared to controls; Er correlated positively with levels of lipid accumulation and LD size and correlated negatively with cell viability[55]. These findings appear to indicate that single liver cell stiffness mainly depends on the size of LDs, while single liver cell morphology depends on the number of LDs.
SINUSOIDAL BIOMECHANICS IN STEATOSIS AND STEATOHEPATITIS
Dynamic mechanical analysis, which is a standard method for the mechanical characterization of biological specimens, has been used to study liver viscosity and elasticity simultaneously in experimental models of diet-induced fatty liver. Using this method, it was shown that the loss modulus G” is significantly higher in mild steatosis (S1) compared to no steatosis (S0), providing evidence that even modest fat accumulation increases viscosity of the liver tissue[56]. At the same time, however, the mathematically derived microchannel flow model predicts that changes in viscoelasticity of fatty liver at the organ level will directly impact sinusoidal flow and pressure as microcirculatory fluid channels of the liver sinusoids embedded in a viscoelastic matrix become increasingly compressed due to steatosis[57] [Figure 2].
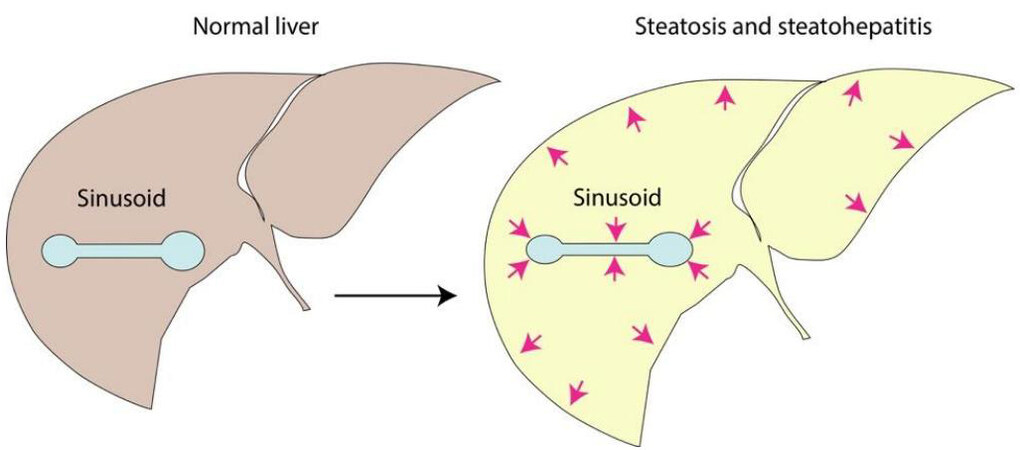
Figure 2. Organ-level biomechanics of the liver in steatosis and steatohepatitis. Schematic illustration of sinusoidal compression from fat accumulation, hepatocellular ballooning and increased interstitial fluid within the confines of the liver capsule, resulting in altered viscoelasticity and increased stress-strain response in the hepatic vascular space.
The development of steatohepatitis represents an additional mechanical perturbation that further complicates biomechanical and rheological modeling in NAFLD[58]. Increasingly inefficient incorporation of fatty acids and glycerolipid derivatives into LDs results in their retention in the ER, promoting ER stress and “overflow” into other lipid pathways, which contributes to steatohepatitis via increased production of ceramides, eicosanoids, and other lipid compounds with toxic effects on the liver[50]. A key morphological manifestation of steatohepatitis, which has a significant impact on sinusoidal biomechanics, is hepatocellular ballooning. Hepatocellular ballooning indicates disruption of the cytoskeleton[59]. Ballooned hepatocytes may double their diameter over their original size and compress the space of Disse, contributing to sinusoidal narrowing and distortion[59]. Hepatocyte enlargement and inflammatory expansion of interstitial fluids exacerbate the volumetric squeeze of the sinusoids within the liver capsule, leading to increased stiffness by the stress-strain response and further reducing sinusoidal space[58]. Calculations from liver elastography in an experimental model of diet-induced fatty liver have demonstrated that hepatocellular ballooning and inflammation are associated with an approximately 20% reduction of the sinusoidal diameter, representing a significant loss of vascular space[58,60]. As such, these viscoelasticity assessments appear consistent with earlier architectural and hemodynamic observations about the effect of steatosis on liver microcirculation, describing tortuous and narrowed channels and impaired sinusoidal flow in experimentally induced fatty liver and human fatty liver[48,49,61,62]. Despite major methodical advances, significant challenges remain in characterizing physiological and pathological changes in blood flow and pressure at the sinusoidal scale[63].
In addition to the impact of external compression, liver sinusoids undergo a number of structural and functional changes in NAFLD, which further disrupt mechanical homeostasis and contribute to disease progression. Sinusoidal endothelial dysfunction develops rapidly in experimentally induced NAFLD[64-66]. LSECs lose their fenestration and build a basal lamina in a process called capillarization[17,67]. The activity of eNOS and the generation of NO is diminished in capillarized LSECs; in the absence of NO, previously quiescent HSCs are activated to a pro-contractile state, leading to sinusoidal vasoconstriction[68]. A major regulator of NO production by LSECs is Kruppel-like factor 2 (KLF2), a mechanosensitive transcription factor that induces genes with anti-inflammatory and anti-fibrotic effects and represses genes encoding for adhesion molecules[68]. Vessel architecture also becomes abnormal in NAFLD. Histologic studies have demonstrated highly disorganized sinusoids with irregular blood vessels and multiple “blebs”, which are thought to represent obstructed sinusoids or leakage from disruption of the normal sinusoidal wall[69].
Capillarized LSECs secrete cytokines and other bioactive substances that recruit inflammatory cells and induce adhesion of portal blood cells to the sinusoidal endothelium, promoting the development of microthrombi and further impeding sinusoidal flow[11,16,28]. LSECs subjected to stretch via chronic sinusoidal congestion activate mechanosensitive pathways (mediated by mechanosensors such as integrins, Notch receptors and PIEZO channels) and release the neutrophil chemotactic chemokine CXCL1, promoting the formation of neutrophil extracellular traps (NETs) and furthering the process of thrombosis[70], which is pivotal for the development of increased intrahepatic vascular resistance as discussed later. Recently, evaluation of human steatotic livers with electron microscopy identified another potential source of sinusoidal obstruction in NAFLD: single-cell steatonecrosis, which refers to a process in which fat extruded from a dying hepatocyte becomes a lipid embolus within the sinusoid channel[71]. In sum, there are a number of cellular and molecular mechanisms that affect sinusoidal biomechanics from the inside and outside, resulting in progressive impairment of hepatic microcirculation from the very early phases of NAFLD onward [Figure 3].
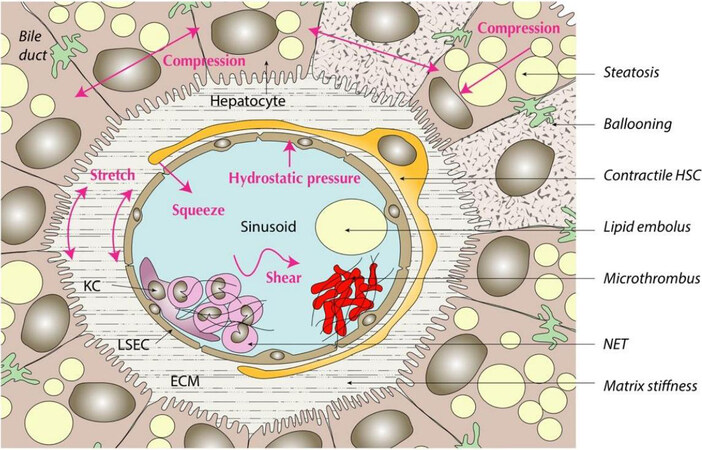
Figure 3. Structural and functional changes in steatosis and steatohepatitis disrupt the mechanical homeostasis of the liver sinusoids; examples of these intravascular and extravascular perturbations are listed on the right. BD: Biliary ductule; ECM: extracellular matrix; HSC: hepatic stellate cell; KC: Kupffer cells; LSEC: liver sinusoidal endothelial cell; NET: neutrophil extracellular trap.
KEY COMPONENTS OF CELLULAR MECHANORESPONSES
Mechanical cues are detected by molecular sensors on both the cell surface and intracellularly (mechanosensing) and transmitted through physical continuum between the contractile cytoskeleton (including actin microfilaments, microtubules, and intermediate filaments) and the nucleoskeleton (mechanotransmission) or via biochemical cascades involving diffusible intermediates (mechanosignaling) to generate a variety of adaptive mechanoresponses[72,73]. Cellular mechanosensors allow the cell to detect physical forces generated within the cell and at interfaces with adjacent cells, ECM, and extracellular fluids[74] [Figure 4]. Deformation of mechanosensors in the plasma membrane and the nuclear membrane, caused by these intracellular and extracellular mechanical forces, may expose new protein binding sites, modulate the activity of transmembrane receptors and associated adhesion molecules, or increase channel/pore conductivity, among other mechanisms[7,75,76].
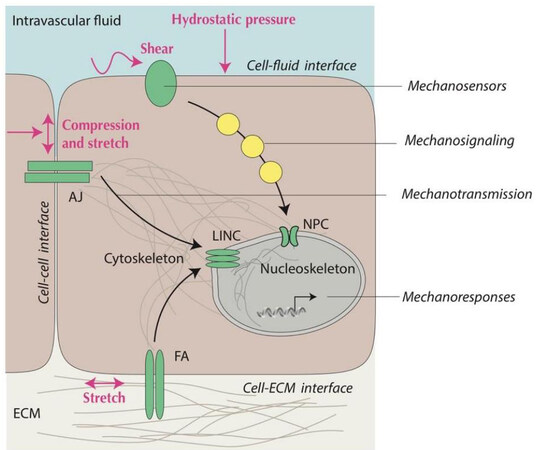
Figure 4. Schematic illustration of mechanical interfaces and components of mechanotransduction. Cells perceive mechanical cues at their interfaces with other cells, ECM, and intravascular (or transcellular) fluids and covert these signals into biochemical and transcriptional information. Mechanosensors (green) at the cell surface (e.g., AJ at the cell-cell interface or FA at the cell-ECM interface) and nuclear membrane (e.g., LINC and NPC) either convey information through the physical connection between the cytoskeleton and nucleoskeleton (mechanotransmission) or convey information through biochemical signaling pathways utilizing enzyme cascades and soluble signaling intermediates (yellow). AJ: Adherens junction; ECM: extracellular matrix; FA: focal adhesion; LINC: linker of nucleoskeleton and cytoskeleton; NPC: nuclear pore complex.
Flow-related physical forces, such as shear stress and hydrostatic pressure, are sensed by various membrane domains (e.g., planar lipid rafts and caveolae, G-protein-coupled receptors, and flow-sensitive ion channels) or specialized cell surface structures (e.g., microvilli and cilia) at the cell-fluid interface, primarily by cells exposed to intravascular fluids (e.g., blood or lymph) or transcellular fluids (e.g., bile)[77]. Downstream effectors of these flow-responsive mechanosensors include biochemical signaling cascades such as MAP kinases, Rho family GTPases, c-Jun amino-terminal kinases, and AKT serine/threonine kinases in addition to soluble bioactive mediators such as NO and intracellular calcium[7]. Major flow-responsive transcription factors, including members of the KLF family (KLF2 and KLF4), Yes-associated protein (YAP), and nuclear factor κB (NFκB), transmit mechanical information to the nucleus as discussed below[76]. Due to their anatomical position, LSECs are the primary sensors of flow and pressure changes in the sinusoidal microcirculation, as these cells are prominently exposed to fluid shear stress and hydrostatic pressure[78].
Adherens junctions anchor adjacent cells to each other and serve as key mechanosensors of crowding and compression at the cell-cell interface in monolayers of epithelial cells (e.g., hepatocytes) and endothelial cells (e.g., LSECs)[79]. Adherens junctions, which are multimolecular structures involving cadherins in the transmembrane portion and multiple adaptor proteins (e.g., catenins) on the cytoplasmic side, establish a physical connection with the cytoskeleton and promote stress fiber formation from actin and myosin[79].
On the other hand, mechanosensing of stiffness and stretching at the interface of cells with ECM involves recognition of matrix proteins (e.g., fibronectin, laminin, and collagen) by specific combinations of integrin heterodimers, which are the key transmembrane receptors of focal adhesions[33,80]. Focal adhesions recruit enzymes (e.g., focal adhesion kinase and Src kinases) to activate downstream effectors, such as the Rho-family GTPases/Rho-associated kinase pathway[5,6]. Similar to adherens junctions, focal adhesions are physically linked to the contractile cytoskeleton by various submembrane protein components, some of which are shared with adherens junctions (e.g., vinculin and talin). The interplay between integrin-based adhesion complexes and cadherin-based adherens junctions is regulated by the contractile cytoskeleton network, which is essential in developing a balanced cellular response to different mechanical cues[81-83].
Integration of cellular mechanoresponses occurs in the nucleus, which receives information from various mechanical interfaces by mechanotransmission and mechanosignaling. Mechanotransmission through the double membrane bilayer of the nuclear envelope occurs through LINC (linker of nucleoskeleton and cytoskeleton), which is a transmembrane complex linked to the actin cytoskeleton on one side and to the nuclear lamina and nuclear matrix on the other side[84,85]. Mechanosignaling to the nucleus involves the import of transcription factors and other macromolecules through the nuclear pore complexes (NPCs), which are formed by the fusion of outer and inner nuclear membranes[86]. There is extensive crosstalk between mechanotransmission and mechanosignaling. For example, stretch-mediated deformation of the nuclear envelope by the contractile cytoskeleton changes the diameter of NPCs[75], which provides a regulatory mechanism for the shuttle of transcriptional regulator molecules between the nucleus and the cytoplasm[86].
YAP and its paralog transcriptional coactivator with PDZ-binding motif (TAZ) have a prominent role in cellular mechanobiology[87,88]. The YAP/TAZ pathway responds to various mechanical cues and to chemical or biological changes, such as hypoxia, inflammation, and energy deprivation[87,89]. Cellular abundance of YAP/TAZ is primarily controlled by proteasomal degradation following phosphorylation by the evolutionarily conserved Hippo kinases mammalian STE20-like (MST) and large tumor suppressor kinase (LATS), while the shuttle of YAP/TAZ between the cytoplasm and nucleus is subject to additional physical and biochemical regulation[90]. Once in the nucleus, binding of YAP/TAZ to the TEAD family of transcription factors regulates the expression of genes with diverse biological effects[90]. While excessive activation of YAP/TAZ has been associated with carcinogenesis[91,92], YAP/TAZ signaling is an essential component of mechanical homeostasis in the liver and other organs[87,88].
MECHANORESPONSES IN LIVER CELLS
As discussed above, the mechanical properties of steatotic and ballooned hepatocytes in NAFLD externally compress the sinusoids[47-49,59]. Moreover, inflammation is associated with expansion of the interstitial fluid and increased stiffness of ECM before significant fibrosis becomes the dominating factor of matrix rigidity[58]. These physical cues trigger mechanoresponses that may contribute to the early stages of NAFLD pathogenesis in many different ways. Even a small increase in ECM rigidity may be perceived by the cell as abnormal biomechanical information, thus stimulating integrin clustering and assembly of focal adhesions to transmit this information to the nucleus via the contractile cytoskeleton and various biochemical signaling cascades[6,26,93]. The importance of cell-ECM interactions in hepatocyte function has been demonstrated in vitro. Culture of hepatocytes on collagen matrix of increasing rigidity leads to progressively impaired metabolic functions (e.g., albumin and glycogen synthesis) and inhibition of the hepatocyte nuclear factor 4α (HNF4α) transcriptional network while strengthening markers of cell proliferation[94]. Notably, HNF4α expression in hepatocytes cultured on stiff matrix was rescued by blocking the Rho/Rho-associated protein kinase pathway, confirming the role of adhesion-associated mechanisms in hepatocellular mechanosensitivity[94].
Similar to hepatocytes, LSECs respond to mechanical cues received from the ECM. Gene ontology analysis of the proteins associated with focal adhesions in primary human LSECs cultured on soft (0.2 kPa) and stiff (32 kPa) collagen-coated gels demonstrated recruitment of phosphofructokinase, one of the rate-limiting glycolytic enzymes, to focal adhesions in LSECs cultured on stiff matrix[95]. In a stiff milieu, researchers found that glycolytic enzymes bind to the cytoskeleton and promote stress fiber formation, changing the conformation of NPCs and thereby allowing NF-κB to access the nuclear chromatin, leading to increased CXCL1 expression and ultimately causing increased sinusoidal pressure and liver fibrosis[95]. Inhibition of glycolysis prevented stiffness-induced angiogenesis in these experiments, corroborating the role of mechanosensitivity in liver disease progression.
Recent research has provided insight into the interplay between hepatocytes and HSCs in regulating the mechanical homeostasis of the liver sinusoids. In the space of Disse, thorn-like projections or spines of HSCs appear to be physically connected to neighboring hepatocytes via adherens junctions[96]. However, as demonstrated in a carbon tetrachloride (CCl4)-induced model of liver injury, adherens junctions between HSCs and hepatocytes vanish in the setting of hepatocellular ballooning, suggesting that an intact cytoskeleton is important in maintaining this connection. Moreover, in this model, HSCs that lost their adherens junctions increasingly displayed a pro-fibrogenic phenotype[96]. These observations suggest that cell-cell contact between HSCs and hepatocytes may be required to keep HSCs quiescent. However, given evidence of crosstalk between cadherin-based adherens junctions and integrin-based focal adhesions[81,82], it is also possible that imbalances at cell-cell and cell-ECM interfaces in this experimental model of NAFLD contribute to the activation of HSCs.
In addition to receiving mechanical cues at the cell-ECM interface, LSECs are continuously exposed to wall stretch and fluid shear stress from sinusoidal blood flow[78]. LSECs and hepatocytes communicate through paracrine interactions across the space of Disse, and these interactions are mechanosensitive, as exposure of LSECs to increasing matrix stiffness and shear stress modulates the phenotype of hepatocytes[97]. It is increasingly recognized that mechanical signals, including changes in sinusoidal blood flow, play a key role in the initiation and termination of liver regeneration[98]. Recent work utilizing a 3-D structural analysis of the liver sinusoidal network following partial hepatectomy found that increased portal blood flow induces acute liver growth; this effect involves mechanosensing of hemodynamic changes by LSECs via G-protein-coupled receptors, intracellular calcium release, and MAPK signaling[99].
Additional aspects of mechanosignaling in the pathogenesis of NAFLD have been recently discussed elsewhere[100]. In the following section, we focus in more detail on an intriguing area of liver mechanobiology: the potential role of mechanical signals related to steatosis and steatohepatitis as contributors to the development of portal hypertension in the pre-cirrhotic liver.
MECHANOBIOLOGY OF EARLY PORTAL HYPERTENSION IN FATTY LIVER
How increased sinusoidal pressure (due to early impediments to hepatic microcirculation as discussed above) may contribute to the complex pathogenesis of NAFLD is not entirely clear, but accumulating evidence suggests that it is an important mechanical cue that contributes to disease progression in the nonfibrotic stage of liver disease[65,101,102]. It is well established that sinusoidal portal hypertension is the cause of major complications in advanced liver disease. Portal hypertension, which is characterized by increased intrahepatic vascular resistance followed by the development of splanchnic dysregulation and porto-systemic collaterals, is defined as a hepatic venous pressure gradient (HVPG) of > 5 mmHg (measured as the difference between wedged and free hepatic venous pressure)[103]. Clinically significant portal hypertension (CSPH), at which point complications such as ascites and varices may develop, is defined as a HVPG of ≥ 10 mmHg[103]. While the significance of subclinical portal hypertension (meaning a HVPG greater than 5 mmHg but smaller than 10 mmHg) in the pathogenesis of NAFLD remains incompletely understood[104], it has been speculated that mild elevations in sinusoidal pressure contribute to the progression of fibrosis in NAFLD, suggesting a bidirectional cause-and-effect relationship between portal hypertension and liver fibrosis[105,106].
Enlarged spleen size appears to be common in NAFLD, even in the absence of cirrhosis[107,108]. It is tempting to speculate that this finding is a manifestation of non-cirrhotic portal hypertension in NAFLD, and indeed, increased spleen stiffness on shear wave elastography has been shown to predict liver fibrosis and portal hypertension[108]. However, given the fact that the spleen is an immune organ, its enlargement in NAFLD may also reflect chronic inflammation and immune activation, although there has been no correlation between spleen size and the severity of NAFLD[108]. Further studies will be required to clarify this matter.
Early studies in diet-induced animal models of fatty liver described decreased portal blood flow, increased sinusoidal narrowing, and increased portal pressure in the setting of simple steatosis as compared to controls[48]. These findings have been corroborated by subsequent work, indicating that portal hypertension occurs in experimental NAFLD without significant inflammation or fibrosis[65,101,102]. It has also been established that increased portal pressure is associated with vascular hyperreactivity of the sinusoidal vessels and increased intrahepatic vascular resistance[65,101]. Consistent with these experimental data, portal hypertension and even CSPH have been reported in cases of NAFLD in which fibrosis is mild or even absent[109,110]. The severity of steatosis may be the only predictor of elevated portal pressure in these clinical scenarios[64,109].
There is also evidence that increased sinusoidal pressure promotes neoangiogenesis in the liver, which is a characteristic feature of portal hypertension and has been implicated in the progression of NAFLD[111]. Elevated serum VEGF concentrations have been found in patients with steatosis and steatohepatitis compared to healthy individuals[112]. In experimentally induced steatosis, VEGF levels are increased within 3 days, indicating that angiogenic factors are activated before any significant fibrosis develops[113]. Vessel perfusion of LSECs in mouse livers perfused ex vivo and mechanical stretching of primary cultured human LSECs in vitro has been shown to activate β1 integrins and VEGF receptor 3, indicating that mechanotransduction alone is sufficient to turn on angiocrine signals, including hepatocyte growth factor (HGF) and pro-inflammatory cytokines, such as interleukin-6 and TNFα[114]. These findings corroborate the notion that increased sinusoidal pressure represents an essential mechanical cue in the early phases of NAFLD and may serve as the impetus for pro-angiogenic and pro-fibrotic mechanisms in the pathogenesis. Further research is needed to identify the precise mechanobiological drivers of the sinusoidal pressure-fibrosis relationship.
CONCLUSION
Steatosis and steatohepatitis are seen in the early stages of NAFLD, at which point fibrosis, the hallmark histological feature associated with increased risk of major liver-related complications such as CSPH, is typically absent. However, it is increasingly recognized that steatosis and steatohepatitis are associated with molecular and cellular changes that promote the development of fibrosis and contribute to disease progression. Accumulating evidence indicates that changes in the biomechanics of liver and disruption of mechanical homeostasis in liver sinusoids during steatosis and steatohepatitis facilitate a pro-angiogenic and pro-fibrotic milieu. Mechanoresponses in hepatocytes and non-parenchymal liver cells, such as LSECs and HSCs, to abnormal or excessive mechanical cues have been observed in a variety of clinical settings and experimental models with implications for adverse outcomes in NAFLD. It is therefore essential to improve our understanding of liver mechanobiology from the earliest stages of NAFLD to help identify novel targets in disease prevention and management.
DECLARATIONS
Authors’ ContributionsConceived the manuscript: Baffy G
Edited and reviewed the final manuscript: Mitten EK, Baffy G
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflict of Interest StatementBoth authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
REFERENCES
1. Iskratsch T, Wolfenson H, Sheetz MP. Appreciating force and shape-the rise of mechanotransduction in cell biology. Nat Rev Mol Cell Biol 2014;15:825-33.
2. Mammoto T, Ingber DE. Mechanical control of tissue and organ development. Development 2010;137:1407-20.
4. Discher DE, Janmey P, Wang YL. Tissue cells feel and respond to the stiffness of their substrate. Science 2005;310:1139-43.
5. DuFort CC, Paszek MJ, Weaver VM. Balancing forces: architectural control of mechanotransduction. Nat Rev Mol Cell Biol 2011;12:308-19.
6. Humphrey JD, Dufresne ER, Schwartz MA. Mechanotransduction and extracellular matrix homeostasis. Nat Rev Mol Cell Biol 2014;15:802-12.
7. Tanaka K, Joshi D, Timalsina S, Schwartz MA. Early events in endothelial flow sensing. Cytoskeleton (Hoboken) 2021;78:217-31.
8. Wang N, Tytell JD, Ingber DE. Mechanotransduction at a distance: mechanically coupling the extracellular matrix with the nucleus. Nat Rev Mol Cell Biol 2009;10:75-82.
9. Belaadi N, Aureille J, Guilluy C. Under pressure: mechanical stress management in the nucleus. Cells 2016;5:27.
13. Mashek DG. Hepatic lipid droplets: a balancing act between energy storage and metabolic dysfunction in NAFLD. Mol Metab 2021;50:101115.
14. Scorletti E, Carr RM. A new perspective on NAFLD: focusing on lipid droplets. J Hepatol 2022;76:934-45.
15. Seebacher F, Zeigerer A, Kory N, Krahmer N. Hepatic lipid droplet homeostasis and fatty liver disease. Semin Cell Dev Biol 2020;108:72-81.
17. Hammoutene A, Rautou PE. Role of liver sinusoidal endothelial cells in nonalcoholic fatty liver disease. J Hepatol 2019;70:1278-91.
18. Long Y, Niu Y, Liang K, Du Y. Mechanical communication in fibrosis progression. Trends Cell Biol 2022;32:70-90.
19. Ma H, Liu X, Zhang M, Niu J. Liver sinusoidal endothelial cells are implicated in multiple fibrotic mechanisms. Mol Biol Rep 2021;48:2803-15.
20. Zhu C, Tabas I, Schwabe RF, Pajvani UB. Maladaptive regeneration - the reawakening of developmental pathways in NASH and fibrosis. Nat Rev Gastroenterol Hepatol 2021;18:131-42.
21. Chen G, Xia B, Fu Q, et al. Matrix mechanics as regulatory factors and therapeutic targets in hepatic fibrosis. Int J Biol Sci 2019;15:2509-21.
22. Park S, Chen Y. Mechanics of biological systems. In. Introduction to mechanobiology and experimental techniques: Morgan & Claypool Publishers; 2019. Available from: https://iopscience.iop.org/book/mono/978-1-64327-392-1.pdf [Last accessed on 16 Mar 2023].
23. Verdier C, Etienne J, Duperray A, Preziosi L. Review: rheological properties of biological materials. Comptes Rendus Physique 2009;10:790-811.
24. Park S, Jung WH, Pittman M, Chen J, Chen Y. The effects of stiffness, fluid viscosity, and geometry of microenvironment in homeostasis, aging, and diseases: a brief review. J Biomech Eng 2020:142.
25. Charrier EE, Pogoda K, Wells RG, Janmey PA. Control of cell morphology and differentiation by substrates with independently tunable elasticity and viscous dissipation. Nat Commun 2018;9:449.
26. Wells RG. The role of matrix stiffness in regulating cell behavior. Hepatology 2008;47:1394-400.
27. Vollmar B, Menger MD. The hepatic microcirculation: mechanistic contributions and therapeutic targets in liver injury and repair. Physiol Rev 2009;89:1269-339.
28. Poisson J, Lemoinne S, Boulanger C, et al. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J Hepatol 2017;66:212-27.
29. Mönkemöller V, Øie C, Hübner W, Huser T, McCourt P. Multimodal super-resolution optical microscopy visualizes the close connection between membrane and the cytoskeleton in liver sinusoidal endothelial cell fenestrations. Sci Rep 2015;5:16279.
30. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev 2008;88:125-72.
31. Baffy G. Kupffer cells in nonalcoholic fatty liver disease: the emerging view. J Hepatol 2009;51:212-23.
32. Cromar GL, Xiong X, Chautard E, Ricard-Blum S, Parkinson J. Toward a systems level view of the ECM and related proteins: a framework for the systematic definition and analysis of biological systems. Proteins 2012;80:1522-44.
33. Duscher D, Maan ZN, Wong VW, et al. Mechanotransduction and fibrosis. J Biomech 2014;47:1997-2005.
34. Bedossa P, Paradis V. Liver extracellular matrix in health and disease. J Pathol 2003;200:504-15.
35. Gong Z, Szczesny SE, Caliari SR, et al. Matching material and cellular timescales maximizes cell spreading on viscoelastic substrates. Proc Natl Acad Sci USA 2018;115:E2686-95.
37. Tamaki N, Kurosaki M, Huang DQ, Loomba R. Noninvasive assessment of liver fibrosis and its clinical significance in nonalcoholic fatty liver disease. Hepatol Res 2022;52:497-507.
38. Sharma AK, Reis J, Oppenheimer DC, et al. Attenuation of shear waves in normal and steatotic livers. Ultrasound Med Biol 2019;45:895-901.
39. Sugimoto K, Moriyasu F, Oshiro H, et al. Clinical utilization of shear wave dispersion imaging in diffuse liver disease. Ultrasonography 2020;39:3-10.
40. Karlas T, Petroff D, Sasso M, et al. Individual patient data meta-analysis of controlled attenuation parameter (CAP) technology for assessing steatosis. J Hepatol 2017;66:1022-30.
41. Poul SS, Parker KJ. Fat and fibrosis as confounding cofactors in viscoelastic measurements of the liver. Phys Med Biol 2021;66:045024.
42. Pearson A, Dujardin PA, d’Alteroche L, et al. Vibration-controlled transient elastography for noninvasive evaluation of liver steatosis. Med Phys 2022;49:1507-21.
43. Parker KJ, Ormachea J. The quantification of liver fat from wave speed and attenuation. Phys Med Biol 2021;66:145011.
45. Shoham N, Girshovitz P, Katzengold R, Shaked NT, Benayahu D, Gefen A. Adipocyte stiffness increases with accumulation of lipid droplets. Biophys J 2014;106:1421-31.
46. Sahini N, Borlak J. Recent insights into the molecular pathophysiology of lipid droplet formation in hepatocytes. Prog Lipid Res 2014;54:86-112.
47. Ijaz S, Yang W, Winslet MC, Seifalian AM. Impairment of hepatic microcirculation in fatty liver. Microcirculation 2003;10:447-56.
48. Wada K, Fujimoto K, Fujikawa Y, Shibayama Y, Mitsui H, Nakata K. Sinusoidal stenosis as the cause of portal hypertension in choline deficient diet induced fatty cirrhosis of the rat liver. Acta Pathol Jpn 1974;24:207-17.
49. Yoshihara H, Hijioka T, Eguchi H, et al. Hepatic microcirculatory disturbance in fatty liver as a cause of portal hypertension. J Gastroenterol Hepatol 1989;4 Suppl 1:279-81.
50. Gluchowski NL, Becuwe M, Walther TC, Farese RV Jr. Lipid droplets and liver disease: from basic biology to clinical implications. Nat Rev Gastroenterol Hepatol 2017;14:343-55.
51. Faulkner CS, White CM, Shah VH, Jophlin LL. A single nucleotide polymorphism of
52. Romero LM, Dickens MJ, Cyr NE. The Reactive Scope Model - a new model integrating homeostasis, allostasis, and stress. Horm Behav 2009;55:375-89.
53. Liu YL, Reeves HL, Burt AD, et al.
54. Li R, Bu Y, Yang C, Wang J. Effects of lipid deposition on viscoelastic response in human hepatic cell line HepG2. Front Physiol 2021;12:684121.
55. Baldini F, Bartolozzi A, Ardito M, et al. Biomechanics of cultured hepatic cells during different steatogenic hits. J Mech Behav Biomed Mater 2019;97:296-305.
56. Zhang X, Gao X, Zhang P, et al. Dynamic mechanical analysis to assess viscoelasticity of liver tissue in a rat model of nonalcoholic fatty liver disease. Med Eng Phys 2017;44:79-86.
58. Parker KJ, Ormachea J, Drage MG, Kim H, Hah Z. The biomechanics of simple steatosis and steatohepatitis. Phys Med Biol 2018;63:105013.
59. Caldwell S, Ikura Y, Dias D, et al. Hepatocellular ballooning in NASH. J Hepatol 2010;53:719-23.
60. Ogawa S, Moriyasu F, Yoshida K, et al. Relationship between liver tissue stiffness and histopathological findings analyzed by shear wave elastography and compression testing in rats with nonalcoholic steatohepatitis. J Med Ultrason (2001) 2016;43:355-60.
61. Seifalian AM, Piasecki C, Agarwal A, Davidson BR. The effect of graded steatosis on flow in the hepatic parenchymal microcirculation. Transplantation 1999;68:780-4.
62. Balci A, Karazincir S, Sumbas H, Oter Y, Egilmez E, Inandi T. Effects of diffuse fatty infiltration of the liver on portal vein flow hemodynamics. J Clin Ultrasound 2008;36:134-40.
63. Li N, Zhang X, Zhou J, et al. Multiscale biomechanics and mechanotransduction from liver fibrosis to cancer. Adv Drug Deliv Rev 2022;188:114448.
64. Francque S, Verrijken A, Mertens I, et al. Noncirrhotic human nonalcoholic fatty liver disease induces portal hypertension in relation to the histological degree of steatosis. Eur J Gastroenterol Hepatol 2010;22:1449-57.
65. Pasarín M, La Mura V, Gracia-Sancho J, et al. Sinusoidal endothelial dysfunction precedes inflammation and fibrosis in a model of NAFLD. PLoS One 2012;7:e32785.
66. Miyao M, Kotani H, Ishida T, et al. Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression. Lab Invest 2015;95:1130-44.
67. Schaffner F, Popper H. Capillarization of hepatic sinusoids in man. Gastroenterology 1963;44:239-42.
68. Shah V, Haddad FG, Garcia-Cardena G, et al. Liver sinusoidal endothelial cells are responsible for nitric oxide modulation of resistance in the hepatic sinusoids. J Clin Invest 1997;100:2923-30.
69. Francque S, Laleman W, Verbeke L, et al. Increased intrahepatic resistance in severe steatosis: endothelial dysfunction, vasoconstrictor overproduction and altered microvascular architecture. Lab Invest 2012;92:1428-39.
70. Hilscher MB, Sehrawat T, Arab JP, et al. Mechanical stretch increases expression of cxcl1 in liver sinusoidal endothelial cells to recruit neutrophils, generate sinusoidal microthombi, and promote portal hypertension. Gastroenterology 2019;157:193-209.e9.
71. Wisse E, Braet F, Shami GJ, et al. Fat causes necrosis and inflammation in parenchymal cells in human steatotic liver. Histochem Cell Biol 2022;157:27-38.
72. Labernadie A, Trepat X. Sticking, steering, squeezing and shearing: cell movements driven by heterotypic mechanical forces. Curr Opin Cell Biol 2018;54:57-65.
74. Vogel V, Sheetz M. Local force and geometry sensing regulate cell functions. Nat Rev Mol Cell Biol 2006;7:265-75.
75. Weis K. Regulating access to the genome: nucleocytoplasmic transport throughout the cell cycle. Cell 2003;112:441-51.
76. Chatterjee S. Endothelial mechanotransduction, redox signaling and the regulation of vascular inflammatory pathways. Front Physiol 2018;9:524.
78. Sun X, Harris EN. New aspects of hepatic endothelial cells in physiology and nonalcoholic fatty liver disease. Am J Physiol Cell Physiol 2020;318:C1200-13.
79. Angulo-Urarte A, van der Wal T, Huveneers S. Cell-cell junctions as sensors and transducers of mechanical forces. Biochim Biophys Acta Biomembr 2020;1862:183316.
80. Zamir E, Geiger B. Molecular complexity and dynamics of cell-matrix adhesions. J Cell Sci 2001;114:3583-90.
81. Wang Y, Jin G, Miao H, Li JY, Usami S, Chien S. Integrins regulate VE-cadherin and catenins: dependence of this regulation on Src, but not on Ras. Proc Natl Acad Sci USA 2006;103:1774-9.
82. Rooij J, Kerstens A, Danuser G, Schwartz MA, Waterman-Storer CM. Integrin-dependent actomyosin contraction regulates epithelial cell scattering. J Cell Biol 2005;171:153-64.
83. Zuidema A, Wang W, Sonnenberg A. Crosstalk between cell adhesion complexes in regulation of mechanotransduction. Bioessays 2020;42:e2000119.
84. Sosa BA, Rothballer A, Kutay U, Schwartz TU. LINC complexes form by binding of three KASH peptides to domain interfaces of trimeric SUN proteins. Cell 2012;149:1035-47.
85. Guilluy C, Osborne LD, Van Landeghem L, et al. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat Cell Biol 2014;16:376-81.
86. Matsuda A, Mofrad MRK. On the nuclear pore complex and its emerging role in cellular mechanotransduction. APL Bioeng 2022;6:011504.
87. Dupont S, Morsut L, Aragona M, et al. Role of YAP/TAZ in mechanotransduction. Nature 2011;474:179-83.
88. Halder G, Dupont S, Piccolo S. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat Rev Mol Cell Biol 2012;13:591-600.
89. Yu FX, Zhao B, Guan KL. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 2015;163:811-28.
90. Mo JS, Yu FX, Gong R, Brown JH, Guan KL. Regulation of the Hippo-YAP pathway by protease-activated receptors (PARs). Genes Dev 2012;26:2138-43.
91. Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the roots of cancer. Cancer Cell 2016;29:783-803.
92. Pocaterra A, Romani P, Dupont S. YAP/TAZ functions and their regulation at a glance. J Cell Sci 2020;133:jcs230425.
93. Martino F, Perestrelo AR, Vinarský V, Pagliari S, Forte G. Cellular mechanotransduction: from tension to function. Front Physiol 2018;9:824.
94. Desai SS, Tung JC, Zhou VX, et al. Physiological ranges of matrix rigidity modulate primary mouse hepatocyte function in part through hepatocyte nuclear factor 4 alpha. Hepatology 2016;64:261-75.
95. Greuter T, Yaqoob U, Gan C, et al. Mechanotransduction-induced glycolysis epigenetically regulates a CXCL1-dominant angiocrine signaling program in liver sinusoidal endothelial cells
96. Urushima H, Yuasa H, Matsubara T, et al. Activation of hepatic stellate cells requires dissociation of e-cadherin-containing adherens junctions with hepatocytes. Am J Pathol 2021;191:438-53.
97. Li W, Li P, Li N, et al. Matrix stiffness and shear stresses modulate hepatocyte functions in a fibrotic liver sinusoidal model. Am J Physiol Gastrointest Liver Physiol 2021;320:G272-82.
98. Song Z, Gupta K, Ng IC, Xing J, Yang YA, Yu H. Mechanosensing in liver regeneration. Semin Cell Dev Biol 2017;71:153-67.
99. Ishikawa J, Takeo M, Iwadate A, et al. Mechanical homeostasis of liver sinusoid is involved in the initiation and termination of liver regeneration. Commun Biol 2021;4:409.
100. Mitten EK, Baffy G. Mechanotransduction in the pathogenesis of nonalcoholic fatty liver disease. J Hepatol 2022;77:1642-56.
101. Van der Graaff D, Kwanten WJ, Couturier FJ, et al. Severe steatosis induces portal hypertension by systemic arterial hyporeactivity and hepatic vasoconstrictor hyperreactivity in rats. Lab Invest 2018;98:1263-75.
102. García-Lezana T, Raurell I, Bravo M, et al. Restoration of a healthy intestinal microbiota normalizes portal hypertension in a rat model of nonalcoholic steatohepatitis. Hepatology 2018;67:1485-98.
103. Bosch J, Iwakiri Y. The portal hypertension syndrome: etiology, classification, relevance, and animal models. Hepatol Int 2018;12:1-10.
106. Baffy G, Bosch J. Overlooked subclinical portal hypertension in non-cirrhotic NAFLD: Is it real and how to measure it? J Hepatol 2022;76:458-63.
107. Tsushima Y, Endo K. Spleen enlargement in patients with nonalcoholic fatty liver: correlation between degree of fatty infiltration in liver and size of spleen. Dig Dis Sci 2000;45:196-200.
108. Tarantino G, Citro V, Balsano C. Liver-spleen axis in nonalcoholic fatty liver disease. Expert Rev Gastroenterol Hepatol 2021;15:759-69.
109. Mendes FD, Suzuki A, Sanderson SO, Lindor KD, Angulo P. Prevalence and indicators of portal hypertension in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol 2012;10:1028-33.e2.
110. Rodrigues SG, Montani M, Guixé-Muntet S, De Gottardi A, Berzigotti A, Bosch J. Patients with signs of advanced liver disease and clinically significant portal hypertension do not necessarily have cirrhosis. Clin Gastroenterol Hepatol 2019;17:2101-2109.e1.
111. Lei L, Ei Mourabit H, Housset C, Cadoret A, Lemoinne S. Role of angiogenesis in the pathogenesis of NAFLD. J Clin Med 2021;10:1338.
112. Coulon S, Francque S, Colle I, et al. Evaluation of inflammatory and angiogenic factors in patients with nonalcoholic fatty liver disease. Cytokine 2012;59:442-9.
113. Coulon S, Legry V, Heindryckx F, et al. Role of vascular endothelial growth factor in the pathophysiology of nonalcoholic steatohepatitis in two rodent models. Hepatology 2013;57:1793-805.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Mitten EK, Baffy G. Mechanobiology in the development and progression of nonalcoholic fatty liver disease: an updated review. Metab Target Organ Damage 2023;3:2. http://dx.doi.org/10.20517/mtod.2022.37
AMA Style
Mitten EK, Baffy G. Mechanobiology in the development and progression of nonalcoholic fatty liver disease: an updated review. Metabolism and Target Organ Damage. 2023; 3(1): 2. http://dx.doi.org/10.20517/mtod.2022.37
Chicago/Turabian Style
Mitten, Emilie K., György Baffy. 2023. "Mechanobiology in the development and progression of nonalcoholic fatty liver disease: an updated review" Metabolism and Target Organ Damage. 3, no.1: 2. http://dx.doi.org/10.20517/mtod.2022.37
ACS Style
Mitten, EK.; Baffy G. Mechanobiology in the development and progression of nonalcoholic fatty liver disease: an updated review. Metab Target Organ Damage. 2023, 3, 2. http://dx.doi.org/10.20517/mtod.2022.37
About This Article
Special Issue
Copyright

Data & Comments
Data



 Cite This Article 15 clicks
Cite This Article 15 clicks

 Like This Article 35
likes
Like This Article 35
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.