
Association of NAFLD/NASH, and MAFLD/MASLD with chronic kidney disease: an updated narrative review
 0
0
Abstract
Chronic kidney disease (CKD) and nonalcoholic fatty liver disease (NAFLD), metabolic dysfunction-associated fatty liver disease (MAFLD) and metabolic dysfunction-associated steatotic liver disease (MASLD) account for substantial financial burden worldwide. These alarming features call for enhanced efforts to prevent and manage the development and progression of CKD. Accumulating evidence supporting a causal role of NAFLD/MAFLD/MASLD-in CKD opens new horizons to achieve this aim. Recent epidemiological studies and meta-analyses exploring the association of NAFLD/MAFLD/MASLD with CKD and the characteristics of NAFLD/MAFLD/MASLD associated with the odds of incident CKD are discussed. The involved pathomechanisms, including the common soil hypothesis, genetics, gut dysbiosis, and portal hypertension, are examined in detail. Finally, lifestyle changes (diet and physical exercise), direct manipulation of gut microbiota, and drug approaches involving statins, renin-angiotensin-aldosterone system inhibitors, GLP-1 Receptor Agonists, Sodium-glucose cotransporter-2, pemafibrate, and vonafexor are examined within the context of prevention and management of CKD among those with NAFLD/MAFLD/MASLD. The evolving NAFLD/MAFLD/MASLD nomenclature may generate confusion among practicing clinicians and investigators. However, comparative studies investigating the pros and contra of different nomenclatures may identify the most useful definitions among NAFLD/MAFLD/MASLD and strategies to identify, prevent, and halt the onset and progression of CKD.
Keywords
INTRODUCTION
History and definitions of NAFLD/MAFLD/MASLD
Nonalcoholic fatty liver disease (NAFLD) nomenclature was modeled in 1986 based on the pioneering definition of nonalcoholic steatohepatitis (NASH), which had been coined in 1980[1]. In 2005, anticipatory suggestions to include the “positive” criterion (i.e., “metabolic”) as opposed to the “negative” diagnosis of exclusion (i.e., “nonalcoholic”)[2] did not encounter any immediate reception. However, in 2020, Mendez-Sanchez et al. endorsed renaming NAFLD to MAFLD (metabolic dysfunction-associated fatty liver disease)[3]. Finally, in 2023, Rinella et al. proposed renaming NAFLD and MAFLD to MASLD (metabolic dysfunction-associated steatotic liver disease)[4].
Table 1 highlights the commonalities and differences between the various definitions: NAFLD/MAFLD/MASLD[5,6]
Commonalities and differences between the various definitions: NAFLD/MAFLD/MASLD
| Essential common requirement | Specific diagnostic criteria | |
| NAFLD | Hepatic steatosis documented histologically, with imaging techniques or biomarkers | Absence of any competing causes of steatosis* |
| MAFLD | Presence of ≥ 1 (out of three) metabolic conditions^ | |
| MASLD | Absence of any competing etiologies of steatosis and presence of ≥ 1 (out of five) metabolic conditions§ |
With this evolving backset, some experts believe that the continuing controversy on the nomenclature of NAFLD is confusing clinical hepatologists[7]. Of concern in this regard is that while NAFLD and MAFLD define different patient populations[8,9], MASLD and NAFLD reportedly describe the same patient population[10].
Chronic kidney disease as a component of the cardiovascular-kidney-metabolic syndrome
Chronic kidney disease (CKD) defines progressive loss of renal function, which may eventually result, among a subset of individuals, in end-stage renal disease (ESRD), where renal replacement is the only way to sustain patients’ life with either dialysis or renal transplantation[11]. Various etiologies may eventually result in CKD exhibiting variable odds of rapid renal disease progression, cardiovascular events, and mortality[12]. However, metabolic syndrome (MetS) undoubtedly is a major etiology predisposing to the initiation and worsening of CKD[13]. Consistently, the recently proposed construct of cardiovascular-kidney-metabolic (CKM) syndrome defines a condition associated with the interconnections linking diabesity, CKD, and cardiovascular disease (CVD)[14]. In the context of the stages of CKM, CKD of moderate-to-high risk positions itself at stage 2, together with other cardiometabolic risk factors [such as hypertriglyceridemia, arterial hypertension (HTN), diabetes, and MetS], suggesting that CKD represents an early target to prevent stages 3 and 4 of the CKM syndrome, which exhibit either preclinical or clinically manifest CVD[14]. Several pathophysiological mechanisms link CKD to CVD development, including shared risk factors (e.g., diabetes and hypertension), perturbed bone mineral metabolism, anemia, volume overload, inflammation, and uremic toxins[15]. As a result, the risk of cardiovascular mortality increases in parallel with decreasing eGFR values[16].
Currently, no approved treatment exists to effectively halt the progression and reverse the dysfunction of CKD, which renders our understanding of the causes and mechanisms of CKD critical to implementing the primary prevention of CKD[17].
Disease burden
Despite important heterogeneity among studies, meta-analytic assessment suggests that the global prevalence of NAFLD, which increased significantly over time, is 32.4% (95%CI: 29.9-34.9)[18]. Overall prevalence and incidence are typically and significantly higher in men than in women[18].
The prevalence of NAFLD and hepatic fibrosis (stages F3-F4) among high-risk groups [(i.e., those with obesity and type 2 diabetes (T2D)] is 75.27% [95%CI: 70.90-79.18] and 6.85% [(95%CI: 3.85-11.90)], respectively, among the obese[19] and 65.04% (95%CI: 61.79-68.15) among those with T2D,
Little is known about the impact of various nomenclatures on the prevalence rates of NAFLD/MAFLD/MASLD. A recent retrospective, cross-sectional study totaling 85,242 adult Chinese reported that MAFLD was more prevalent than NAFLD, that different clinical features characterized MAFLD and NAFLD populations, and that CKD was associated with MAFLD[7]. In contrast, a study from Brazil reported similar prevalence rates and disease risk factors, irrespective of the NAFLD/MAFLD/MASLD nomenclature used[26].
An estimated > 800 million individuals globally, namely > 10% of the general population worldwide, have CKD, with older people, women, racial minorities, individuals with dysmetabolic traits, and developing countries being exposed to higher CKD risk[27,28].
Mendelian-randomization (MR) analysis has identified body mass index, HTN, high-density lipoprotein (HDL) cholesterol, apolipoprotein A-I, lipoprotein(a), T2D, and nephrolithiasis as the variables causally associated with CKD in Europeans[29]. Additionally, a prospective, population-based cohort study conducted on 34,831 individuals reported that hyperuricemia was a significant risk factor for incident CKD after a median 4.1-year follow-up[30]. Collectively, the above-mentioned metabolic risk factors are widely acknowledged to be associated with NAFLD/MAFLD/MASLD[31-33], raising the rational expectation that NAFLD/MAFLD/MASLD and CKD are likely to occur in association.
Genetic cofactors may double the odds of CKD in people of African ancestry rather than among those of European ancestry[34]. These include sickle cell anemia and 2 APOLI polymorphisms.
One of the emerging top causes of mortality worldwide, CKD accounts for approximately 1.2 million deaths and 28 million years of life lost annually[35], being among the few non-communicable diseases that have shown an increased toll of mortality over the past 20 years[27]. Age-adjusted rates support the notion that, in the last two decades, CKD was among the fastest-growing causes of death and CKD is projected to become the fifth cause of mortality by 2040[27,36]. Average healthcare costs are almost three-fold higher among CKD patients than in the average health population and vary incrementally according to stages 3 and 4 of CKD and dialysis[37].
These alarming features call for enhanced efforts to prevent and manage onset and deterioration of CKD and evidence supporting a role of NAFLD/MAFLD/MASLD in the determinism of CKD opens new horizons to achieve this aim.
Aims
The above-summarized scenario of confusing nomenclature changes regarding NAFLD/MAFLD/MASLD and high disease burden (NAFLD/MAFLD/MASLD and CKD), together with accumulating novel data on the epidemiological associations and pathophysiological interconnects among NAFLD/MAFLD/MASLD and CKD, have prompted me to update synthesis and comment of new literature data compared to the principal studies published in 2022 and 2023 on the same topic[38-43]. A distinguishing feature of the present article is its focus on research perspectives.
Strategy of bibliographic research
The basic strategy followed to retrieve those articles cited in my review was to examine the PubMed database on the following query: (((NAFLD[Title/Abstract]) AND (CKD[Title/Abstract])) OR (MAFLD[Title/Abstract])) OR (MASLD[Title/Abstract]). This research, conducted on November 9th, 2023, yielded 1,772 results. Among these, the most recent studies were selected. Additional queries including more specific keywords, such as “epidemiology”, “pathophysiology”, “mendelian-randomization”, “cross-sectional”, “follow-up”, and “management” were utilized as appropriate.
EPIDEMIOLOGY
Epidemiological studies should answer research questions addressing the association linking NAFLD/MAFLD/MASLD in cross-sectional and follow-up investigations and identify the specific risk factors affecting the risk of incident CKD among individuals with NAFLD/MAFLD/MASLD.
Are NAFLD/MAFLD/MASLD associated with CKD in cross-sectional studies?
Over the last few years, cross-sectional studies and one meta-analysis of cross-sectional studies have evaluated the association between NAFLD/MAFLD/MASLD and CKD, such as summarized in Table 2[44-50].
Recent cross-sectional studies and meta-analysis of cross-sectional studies exploring the association of NAFLD/MAFLD/MASLD with CKD[44-50]
| Author (year) | Findings | Comment | |
| Musso et al. (2014)[44] | Meta-analysis of 33 studies totaling 63,902 individuals CKD was defined as persistent eGFR < 60 mL/min/1.73 m2 creatinine clearance < 60 mL/min per 1.73 m2 persistent proteinuria other abnormalities detected by electrolyte or urinary sediment alterations, histology, or imaging history of kidney transplantation | Risk of prevalent CKD was increased among those with NAFLD (OR 2.12, 95%CI: 1.69-2.66) | This meta-analysis also contains an estimate of the risk of incident CKD, as shown in Table 3 |
| Liu et al. (2019)[45] | Taiwan. 37,825 individuals Steatosis assessed with US | At multivariate analysis, individuals with moderate to severe NAFLD were at higher risk of CKD (OR, 1.17, 95%CI: 1.03-1.33) than non-NAFLD subjects | This study is exposed to the risk selection bias given that subjects undergoing health check-up investigations may not represent the general population |
| Akahane et al. (2020)[46] | Japan. 1097 NAFLD and 1097 PS-matched subjects without NAFLD. Steatosis was assessed with US CKD defined as eGFR < 60 mL/min/1.73 m2 | After multivariate adjustment for metabolic confounders, the risks of abnormal albuminuria [OR (95%CI): 1.68 (1.21-2.33), P < 0.01] and CKD [OR (95%CI): 1.54 (1.14-2.07), were increased by 68% and 54%, respectively, per one SD increase in IHTG content (P < 0.01)] | At LRA obesity, HTN, and HUA (but not NAFLD) independently predicted CKD and, among individuals with NAFLD, obesity, HTN, and HIA independently predicted CKD supporting the notion that common CMRFs may mediate the association of NAFKD with CKD |
| Deng et al. (2021)[47] | USA. 1983 subjects with MAFLD (LUSTE) and 1983 PS-matched subjects without MAFLD CKD was defined as either eGFR ≤ 60 mL/min/1.73 m2 or the presence of albuminuria | MAFLD was not independently associated with CKD after PSM | Although PS yielded negative findings, in the patient population of 4,869 subjects from the NHANES 2017-2018 cohort, of whom 1,032 (21.2%) individuals had CKD, a higher prevalence of CKD was observed among MAFLD subjects compared to non-MALFD subjects |
| Sun et al. (2021)[48] | USA. 12,571 individuals from the 3rd NHANES (1988-1994) were included in the analysis. CKD was defined as either CKD stage ≥ 1 or stage ≥ 3) or abnormal albuminuria (urinary albumin-to-creatinine ratio ≥ 3 mg/mmol) | Compared to NAFLD, MAFLD subjects had a higher prevalence of CKD (29.60% vs. 26.56%, P < 0.05) and the prevalence of CKD was higher in MAFLD than in subjects who had “non-MAFLD NAFLD” | In this study, MAFLD captures CKD better than NAFLD |
| Su et al. (2022)[49] | China. 5,594 participants were enrolled CKD was defined as eGFR < 60 mL/min/1.73 m2 or the presence of albuminuria | MAFLD exhibited a higher prevalence of CKD than non-MAFLD controls (16.2% vs. 7.6%, P < 0.001) MAFLD was strongly associated with an increased risk of CKD (OR: 1.35, 95%CI: 1.09-1.67) MAFLD-T2D subtype exhibited a higher risk of CKD (OR: 2.85, 95%CI: 2.24-3.63) Worsening of glucose tolerance in MAFLD was associated with an increased risk of CKD in a dose-dependent manner (P-trend < 0.001), and conversely, good metabolic control in MAFLD was associated with decreased odds of CKD | The strong association of MAFLD with CKD risk is driven by T2D |
| Agustanti et al. (2023)[50] | Meta-analytic review of 11 studies totaling 355,886 individuals CKD was defined as eGFR <60 mL/min/1.73 m2 | MAFLD was associated with a significantly higher prevalence of CKD [OR 1.50, 95%CI: (1.02-2.23); MAFLD and NAFLD patients had a similar prevalence rate of CKD | Conlicting with the study by Sun et al.[48], according to this study both NAFLD and MAFLD identify the same prevalence of CKD |
Studies summarized in Table 2 have yielded discrepant or conflicting findings, which probably occur owing to limited patient populations and may be explained by variable strength of association with CKD of NAFLD vs. MAFLD. Of interest, the two meta-analytic reviews by Musso et al. and Agustanti et al. found that NAFLD and MAFLD, respectively, were associated with a significantly higher prevalence of CKD[44,50]. In this regard, a study conducted among 12,571 individuals from the 3rd National Health and Nutrition Examination Survey (1988-1994) found that MAFLD identifies patients with CKD better than NAFLD[48]. However, according to the meta-analysis conducted by Agustanti, the prevalence of CKD did not vary between MAFLD and NAFLD patients[50].
Finally, it must be pinpointed that cross-sectional studies (and meta-analytic reviews of such studies) cannot ascertain the time frame of disease development, leaving the question fully open as to the typical chicken-or-egg debate: does CKD cause NAFLD/MAFLD/MASLD or vice-versa? Therefore, additional studies should more clearly define whether NAFLD/NASH, MAFLD/MASLD accurately capture prevalent CKD. At any rate, researchers have correctly focused on incident CKD as a model consistent with the notion that, during follow-up, individuals with NAFLD/MAFLD/MASLD are prone to the risk of developing CKD that is not present at the baseline observation.
Are NAFLD/MAFLD/MASLD associated with incident CKD?
The abundance of original studies addressing this research question has justified several meta-analytic reviews over time. In the previously cited study, Agustanti et al. found that the odds of incident CKD were increased among patients with MAFLD [adjusted HR 1.35, 95%CI: (1.18-1.52); test for overall effect
Comparison of published meta-analytic reviews associating incident CKD among individuals with NAFLD/MAFLD/MASLD[44,50,51,52]
| Author (year) | Musso et al. (2014)[44] | Mantovani et al. (2018)[51] | Mantovani et al. (2022)[52] | Agustanti et al. (2023)[50] |
| N. of studies | 33 | 9 | 13 | 11 |
| Enrollees | 63,902 | 96,595 | 1,222,032 | 355,886 subjects |
| CKD definition | persistent eGFR < 60 mL/min/1.73 m2 creatinine clearance < 60 mL/min per 1.73 m2 persistent proteinuria other abnormalities detected by electrolyte or urinary sediment alterations, histology, or imaging history of kidney transplantation | eGFR< 60 mL/min/1.73 m2, with or without overt proteinuria | eGFR < 60 mL/min/1.73 m2, with or without overt proteinuria | eGFR < 60 mL/min/1.73 m2 |
| Assessment of NAFLD severity | NASH or advanced fibrosis | - serum liver enzymes, - FLI or hepatic US scanning | - raised GGT; or -histological fibrosis and/or NFS | NAFLD fibrosis score |
| Years of follow-up | 3-27 | median 5.2 | median 9.7 | 4.6-6.5 |
| Estimated risk of incident CKD risk | HR = 1.79, 95%CI: 1.65-1.95 | HR = 1.37, 95%CI: 1.20-1.53; I2 = 33.5% | HR 1.43, 95%CI: 1.33 to 1.54; I2 = 60.7% | aHR 1.35, 95%CI: [1.18-1.52]; test for overall effect Z = 15.47, |
| Determinants of CKD risk | The severity of NAFLD (namely NASH or advanced fibrosis) was directly associated with CKD stages | "More severe" NAFLD was associated with a higher risk of incident CKD | The odds of incident CKD stage ≥ 3 were greater among subjects with advanced fibrosis | Significant liver fibrosis (but not steatosis) and "more severe MAFLD" were associated with a higher risk of incident CKD |
Do the nomenclatures and the severity of NAFLD/MAFLD/MASLD define the risk of CKD more accurately?
Preliminarily, it should be acknowledged that not all NAFLD/MAFLD/MASLD are alike as regards the risk of incident CKD. Indeed, several studies have found that MAFLD predicts CKD better than NAFLD[48,54-57].
Additionally, Table 3 also identifies the “severity” of NAFLD/MAFLD/MASLD as a determinant of the risk of incident CKD. However, disease severity is defined differently across the four meta-analytic reviews, mirroring variable criteria followed by the individual original studies included in the meta-analyses. Therefore, to gain additional insight into this key topic, some specific studies are summarized in Table 4.
| Author (year) | Series and method | Findings | Conclusion |
| Pan et al. (2015)[58] | 485 participants out of 1,068 obese individuals were submitted to 1H-MRS for the assessment of IFC | The risk of abnormal albuminuria and CKD increased by 68% [OR (95%CI): 1.68 (1.21-2.33), P < 0.01] and 54% [OR (95%CI): 1.54 (1.14-2.07), P < 0.01], respectively, per one SD increase in IHTG content irrespective of age, BMI, and HTN | The severity of steatosis, assessed with IHTG content, is independently associated with CKD in obese adults |
| Zuo et al. (2021)[59] | Community-based prospective study of individuals aged ≥ 40 years and free of CKD at baseline. Mean follow-up 4.4 years CKD was defined as UACR ≥ 30 mg/g, or eGFR ≤ 60 mL/min/1.73 m2 | Incident NAFLD, compared to non-NAFLD, was associated with a higher risk of incident CKD after adjustments for confounding factors Among 534 participants with persistent NAFLD, compared to stable fibrosis, fibrosis progression from low NFS to intermediate/high NFS was associated with an increased risk of incident CKD | Incident NAFLD and worsening of liver fibrosis are associated with higher odds of incident CKD |
| Ciardullo et al. (2022)[60] | Meta-analysis of 7 cross-sectional studies (3 studies conducted in Asia, 3 in Europe, and 1 in the US) totaling 7,736 individuals aged 42 to 69 years | The risk of CKD was higher in patients with LS assessed by VCTE, compared to individuals without LS Elevated LS was also associated with an increased risk of UACR (OR 1.98 95%CI: 1.29-3.05) | This study provides meta-analytic evidence that, among NAFLD patients, high LS is associated with an increased risk of CKD |
| Seo et al. (2022)[61] | longitudinal cohort study of 3,188 T2D patients with normal renal function followed for 8.3 ± 3.6 years. In NAFLD, advanced liver fibrosis was defined as a FIB-4 index ≥ 2.67 CKD was defined as an eGFR of < 60 L/min/1.73 m2 for two consecutive times during follow-up visits | Compared to the non-NAFLD controls, the NAFLD group did not have any higher risk of incident CKD, but among NAFLD patients, advanced liver fibrosis was associated with an increased risk of CKD | Advanced hepatic fibrosis is a risk factor for incident CKD among NAFLD individuals with T2D |
| Sun et al. (2022)[62] | Cross-sectional study comprising 13,915 participants to whom 1,734 additional individuals who had been followed annually for 5 years were added retrospectively CKD was defined as either kidney damage (i.e., pathologic abnormalities or markers of damage, including abnormalities in blood or urine tests or imaging results) or eGFR < 60 mL/min/1.73 m2 for ≥ 3 months | At Cox regression analysis, FIB-4 intermediate risk and high risk significantly predicted CKD. However, only NFS high risk was a significant predictor | FIB-4 and NFS, surrogate indices of hepatic fibrosis, significantly predict CKD |
| Chung et al. (2023)[63] | Utilizing population-based observational data from the KNHIS, a cohort of 1,900,598 T2D patients were followed for a median period of 7.2 years CKD was defined as eGFR < 60 mL/min/1.73 m2 | After adjustment for confounders, individuals with high FLI scores (compared to those with FLI < 30) were associated with a higher risk of ESRD The association between FLI ≥ 60 and incident ESRD was more prominent in women (HR 1.835; 95%CI: 1.689-1.995) than in men (HR 1.106; FLI scores ≥ 60 were associated with higher odds of ESRD in patients with baseline CKD | High FLI scores are associated with higher odds of ESRD among individuals with T2D who have baseline CKD |
Analysis of Table 4 confirms that non-invasive (surrogate) indices of liver fibrosis, such as FIB-4, NFS, and liver stiffness, are consistently associated with the odds of CKD. This is clinically relevant as it enables the identification of the cohort of subjects with NAFLD/MAFLD/MASLD who are more at risk of having incident CKD. However, from a conceptual point of view, epidemiological studies, while proving associations, do not demonstrate causality and, importantly, do not explain the underlying pathomechanisms.
PATHOMECHANISMS
In principle, different scenarios can be envisaged. The hypothesis that pre-existing CKD facilitates the development of incident NAFLD/MAFLD/MASLD is confuted by the consistent body of evidence discussed under point 1.4 above. Therefore, it seems more likely that either NAFLD/MAFLD/MASLD and CKD result from a shared common precursor or that the severity of liver disease owing to NAFLD/MAFLD/MASLD affects the development of incident CKD. However, the “common precursor” hypothesis does not necessarily rule out the direct responsibility of more severe forms of hepatic fibrosis in the development of incident CKD. In other words, multiple mechanisms may variably interact in the individual patient, therefore contributing to the clinical heterogeneity of the disease, which, in close analogy to what occurs for the MetS[64], is a universally acknowledged feature of NAFLD/MAFLD/MASLD in humans[64-72].
Genetics
Associations of specific genes with NAFLD/MAFLD/MASLD in humans contribute to promoting our understanding of disease pathobiology, identifying promising drug targets, and developing polygenic risk scores, which may assist in defining accurate risk stratification in this arena[73,74]. The principal genes which have been strongly associated with the course of NAFLD/MAFLD/MASLD by confirmative studies include PNPLA3, TM6SF2, MBOAT7, GCKR, and HSD17B13[68,73].
Table 5 summarizes some of the most recent studies addressing the impact of genetic variants commonly associated with initiation and worsening of NAFLD/MAFLD/MASLD and CKD[75-78].
Principal studies addressing the role of genetic polymorphisms in relation to CKD in NAFLD/MAFLD/MASLD[75-78]
| Author (year) | Series and method | Findings | Conclusion |
| Sun et al. (2020)[75] | 217 subjects with NAFLD demonstrated histologically CKD defined as any CKD stage from 1 to 5 according to the National Kidney Foundation 2002 clinical practice guidelines | PNPLA3 GG genotype was associated with the risk of CKD and abnormal albuminuria irrespective of conventional risk factors for CKD and severity of NAFLD histology | PNPLA3 genotyping may identify NAFLD patients at higher risk of RTI |
| Mantovani et al. (2020)[76] | 157 T2D patients were submitted to non-invasive assessment with US and VCTE for NAFLD diagnosis. CKD was defined as eGFR < 60 mL/min/1.73 m2 and/or abnormal albuminuria Moreover, PNPLA3 mRNA expression in human tissues, PNPLA3 mRNA and protein expression levels in human cell lines represented in the kidney and the liver were also evaluated | I148M homozygosity was associated with significantly lower e-GFR levels and a higher risk of CKD was independent of LSM ≥ 7 kPa and other risk factors. PNPLA3 mRNA expression was greatest in liver and renal cortex, and podocytes showed high PNPLA3 mRNA and protein levels, similar to those of hepatocytes and hepatic stellate cells, respectively | PNPLA3 I148M was associated with CKD, irrespective of common risk factors of CKD and NAFLD severity PNPLA3 expression levels were especially elevated in renal podocytes |
| Akuta et al. (2021)[77] | A retrospective analysis of the incidence of CVD, extra-hepatic malignancy, and LRE was conducted in 477 Japanese adults with histologically diagnosed NAFLD, with a median follow-up period of 5.9 years | Multivariate analyses established that the three independent predictors of CVD risk were: (1) PNPLA3 genotype; (2) CKD; and (3) FIB-4 index | An interaction among PNPLA3 genotype, CKD, and liver fibrosis collectively determine the risk of CVD in NAFLD |
| Mantovani et al. (2023)[78] | 1,144 middle-aged individuals were recruited. In a subgroup of 144 subjects, the effect of PNPLA3 p.I148M on eGFR was assessed during a median follow-up of 17 months | The p.I148M variant was associated with lower eGFR independent of confounding factors* Prospectively, the p.I148M variant was strongly associated with faster eGFRCKD-EPI decline | The PNPLA3 p.I148M variant carries a detrimental impact on renal function in middle-aged dysmetabolic individuals independent of established risk factors for CKD |
An exhaustive analysis of similar studies is out of the ambit of this review and has recently been published elsewhere[79]. Taken collectively, those studies summarized in Table 5 support a major role of genetics in the development of incident CKD among those with NAFLD/MAFLD/MASLD. In particular, the PNPLA3 p.I148M variant has been associated with a detrimental impact on renal function. Although the pathomechanics underlying this association remains incompletely elucidated, it is possible that the PNPLA3 p.I148M variant exerts this deleterious influence via a mechanism unrelated to the liver, such as shown by the finding that renal podocytes express particularly high levels of PNPLA3 mRNA[78]. These findings are compatible with the notion that the PNPLA3 p.I148M variant could predispose to the development of “fatty kidney disease”, eventually carrying deleterious effects on renal function over time. Interestingly, these deleterious renal outcomes occur irrespective of values of liver stiffness assessed with transient elastography, suggesting that, at least in some cases, the liver could be an innocent bystander of primarily genetic progressive decline in renal function[79]. Therefore, the results of this line of research are closely reminiscent of the concept of two different types of NAFLD (i.e., PNPLA3-related “genetic” and “metabolic”) with variable disease outcomes[80].
Common dysmetabolic soil
Epidemiological evidence
It is widely known that HTN leads to glomerulosclerosis and mild proteinuria independent of dyslipidemia and central obesity[81]. Moreover, up to half of those living with diabetes have diabetic nephropathy (DN), a clinically heterogeneous syndrome featuring persistent albuminuria and progressively declining renal function[82]. DN poses a major healthcare challenge, being a major cause of end-stage kidney disease (ESKD) requiring replacement therapy and carrying the risks of significantly increased cardiovascular morbidity and mortality[82]. In addition to diabetes, obesity also poses formidable risks to renal health. A meta-analytic review of 8 prospective cohort studies, totaling nearly 5 million participants followed for a 3 to 14-year median period, found progressively incremental risks of incident CKD among those with metabolically healthy obesity, metabolically unhealthy normal weight, and metabolically unhealthy obesity (all compared to the metabolically healthy normal-weight) with HR [CI] of 1.41 [1.07-1.74], 1.50 [1.40-1.60], and
Shared pathomechanisms
Experimental and clinical investigations have shown that the MetS is a major player in the development of CKD and that, as articulated in Section 5 of the present review, this relationship is bi-directional, given that the kidneys participate in the homeostasis of glucose and lipids[84].
Metabolic dysfunction exhibits a background of low-grade subclinical inflammation, increased oxidative stress, and upregulated synthesis of multiple profibrotic growth factors[85]. Triggered by insulin resistance and compensatory hyperinsulinemia, multiple pathomechanisms may sustain the development of incident CKD among those with established MetS at the baseline. In addition to the above-mentioned pathomechanisms, these comprise endoplasmic reticulum stress, glomerular hyperfiltration, endothelial dysfunction, activation of the renin-angiotensin system, proliferation of mesangial cells, and expansion of the extracellular matrix[84]. Chronic inflammation contributes to the decrement of GFR characterizing CKD, and inflammation and metabolism are two main pathways leading to CKD progression, with Nrf2 playing the role of the hub[86]. Additionally, SREPB is a key nuclear receptor that controls multiple cellular signals to integrate lipogenesis, endoplasmic reticulum (ER) stress, inflammation, autophagy, and apoptosis, serving a pivotal role in the development of CKD and translating metabolic triggers with inflammatory responses[87].
Taken collectively, the above changes will eventually culminate in the development of microalbuminuria, renal fibrosis, and CKD[84]. Mesangial cells physiologically play a key angiogenic role in glomerular capillary loop development and support the division of a single capillary into multiple loops. Mesangiolysis, featuring loss of injured mesangial cells, occurs in the setting of various cardiometabolic conditions, such as hypertension and diabetes[88]. Besides the more general pathomechanisms summarized above, the individual components of the MetS may damage the kidneys’ health via specific deleterious mechanisms. Among these, HTN, diabetes, and obesity are the best characterized and will be briefly discussed below.
Hypertensive nephropathy
Hypertensive nephropathy (HN) involving hyalinization and sclerosis of interlobular and afferent arterioles, together with fibrosis of glomerular and tubulointerstitial compartments, ranks second after diabetes among the most common causes of ESRD[89]. For years attributed to damaged afferent arterioles and glomeruli mediated by the activation of the renin-angiotensin system (RAS), more recently, HN has been found to result from injured tubular cells, leading to tubulointerstitial fibrosis via epithelial-mesenchymal transition (EMT)[89]. HTN-induced injury of glomeruli damages (post-glomerular) peritubular capillaries, which, in turn, triggers a pathogenic cascade involving hypoxia from endothelial damage and dysfunctional microvasculature, chronic inflammation, eventually leading to fibrosis development owing to dedifferentiation of epithelial cells and EMT; prominent features of HN comprise effacement and loss of podocytes culminating in the disruption of the filtration barrier[89].
Analysis of proteome profiles has provided novel highlights on proteasome-mediated protein degradation, organization of actin cytoskeleton, and Rho GTPase signaling pathway in renal sub-compartments. Data showing that major features in the pathogenesis of HN include alteration of homeostasis of oxygen and energy, as well as of metabolism of amino acid and purines, support the innovative theory that HN can be considered an “acquired error of metabolism”[90].
Diabetic nephropathy
Diabetic nephropathy (DN) exhibits glomerular hypertrophy and glomerulosclerosis, expansion of mesangium, tubulointerstitial inflammatory and fibrotic changes, and loss of podocytes[91].
Chronic hyperglycemia is a key determinant in the pathogenic cascade leading to DN via increased production of advanced glycation end-products. In this setting, glomerular hyperfiltration induces intraglomerular hypertension; moreover, adipokines microinflammation, podocyte depletion, proteinuria, and focal segmental glomerulosclerosis will eventually lead to interstitial fibrosis and expansion of the extracellular matrix[92]. Other pathogenic mechanisms comprise intracellular mesangial cell accumulation of triglyceride and cholesterol ester owing to chronic exposure to insulin-like growth factor-1 (IGF-1), which makes these mesangial cells morphologically similar to foam cells and functionally incapable of responding to migratory and contractile stimuli[93].
Farnesoid X Receptor (FXR), a bile acid sensor that modulates enterohepatic circulation of bile acids, also serves as a master regulator of glucose-lipidic and energy homeostasis, participates in renal reabsorption of water, and is involved in the development of CKD[94]. Studies support the notion that, by improving the renal storage of lipids, glucose homeostasis, renal inflammation, and fibrosis, FXR agonists may prevent DN[94]. In this context, it comes of interest that obeticholic acid (OCA) has exhibited anti-inflammatory and anti-fibrotic properties in the kidneys and the liver in mouse and rat models[95,96]. However, the potentially beneficial outcomes of first-generation FXR agonists including OCA, tropifexor, cilofexor, and nidufexor are typically counterbalanced by HDL-cholesterol lowering, increased LDL-C, and dose-dependent pruritus, which can lead to treatment discontinuation in up 10% of patients[97].
Obesity-related nephropathy
Obesity-related nephropathy (ORN) is sustained by morpho-functional changes occurring among mesangial cells, podocytes, and proximal tubular cells because of impaired renal metabolism of lipids[94].
The condition of obesity itself carries a state of mitochondrial dysfunction and energy depletion[98]. Additional pathogenic features of nephropathy in the obese include increased GFR and renal plasma flow, increased filtration fraction and Na+ tubular reabsorption; this would lead to increased fluid shear stress on podocytes, phenomena of maladaptive renal hypertrophy, detachment of podocytes and, finally, global glomerulosclerosis[98]. Importantly, innovative super-resolution ultrasound Imaging techniques identify structural alterations in the renal vasculature[99].
Dyslipidemic nephropathy
There is a continuous debate about the importance of lipid metabolism in CKD. The concern of low cholesterol levels that could mark cachexia and protein/energy wasting is a debated topic. Hashemi et al., based on an assessment of a cohort of 1,972,851 middle-aged United States veterans, predominantly male, whose serum low-density lipoprotein (LDL) values were available between 2004 and 2006, found that the associations of LDL with mortality and hospitalizations owing to both atherosclerotic and non-atherosclerotic CVD are modulated by the stages of CKD[100]. Conversely, high-density lipoprotein (HDL) cholesterol fractions are deemed to be involved in the development of CKD. Baragetti et al. enrolled 176 individuals and followed them for up to 84 months. This investigation found that low serum values of HDL-cholesterol are associated with a poor prognosis; moreover, the functionality of HDL particles is also impaired among those with impaired renal function, supporting the notion that HDL is associated with the worsening of CKD[101]. A more recent investigation conducted by the same group of investigators in a cohort of 164 CKD patients[102] found that reduced plasma lecithin:cholesterol acyltransferase concentration anticipates the worsening of CKD over time among individuals with baseline renal dysfunction, as well as in the general population. Collectively, the studies discussed above support the notion that lipidemic values are not “innocent bystanders” in the setting of CKD progression, but instead, they actively participate in the decline over time in a fraction of individuals.
Critical steps in the development of CKD among those with atherogenic dyslipidemia include increased expression and activity of SREPB which not only mediates renal lipotoxicity, defined as the accumulation of lipids (i.e., ceramides, diglycerides) capable of inducing cell damage[103] but is also a profibrotic mediator of CKD by directly activating TGF-β via lipid-dependent and -independent pathways[87].
Gut microbiota
Six studies using Mendelian Randomization have established a cause-and-effect association between gut microbiota and CKD [Table 6][17,104-108].
Published Mendelian Randomization studies supporting an association between gut microbiota and CKD[17,104-108]
| Author (year) | Method | Findings | Conclusion |
| Jia et al. (2019)[104] | Genetic variants were instrumented to assess causal associations CKD was defined as eGFR < 60 mL/min/1.73m2 | T2D and CKD were causally associated with higher TMAO levels | This study supports the notion that T2D and CKD increase TMAO levels |
| Mazidi et al. (2020)[105] | MR was conducted using summary-level data from GWAS on microbiota genera, CKD, and parameters of renal function CKD defined as eGFR < 60 mL/min/1.73 m2 | Higher abundance of Desulfovibrio spp. Associated with significantly lower levels of eGFR; these findings were also noted among observed in nondiabetic individuals The Anaerostipes genus was associated with higher eGFR in the overall population and among those non-DM individuals, while it had a non-significant association with the risk of CKD and eGFR among individuals with DM | eGFR is adversely associated with Desulfovibrio spp; and beneficially associated with Anaerostipes spp |
| Luo et al. (2022)[106] | Two-sample MR analysis was performed to assess gut microbiota and metabolites in possible causal relation with 11 cardio-nephrological outcomes CKD was defined as eGFR < 60 mL/min/1.73 m2 | The RR of CKD increased by 7.1% for every 1-unit increased Candida concentration | This study suggests novel mechanisms underlying CKD that are amenable to the use of microbiome- and microbiome-dependent metabolite interventions for its prevention |
| Li et al. (2023)[107] | Two-sample MR analysis of 211 microbiotas and six clinical phenotypes | Class Bacteroidia had a strong causality with lower eGFR after the Bonferroni-corrected test, whereas phylum Actinobacteria was strongly and causally associated with dialysis | This study identifies the specific intestinal flora causally related to the initiation and worsening of CKD at the level of gene prediction |
| Gagnon et al. (2023)[108] | 2-Sample MR 10 metabolites of intestinal origin and 57 microbial taxa abundance were assessed as exposures. Various cardiometabolic health outcomes were assessed, including GFR | 4/7 effect sizes were small. The two largest exposure-outcome effects were markedly attenuated upon inclusion in multivariable MR analyses of BMI or alcohol intake | Findings reject a strong causal impact of human gut microbiota features on cardiometabolic traits, chronic diseases, or longevity. Data suggest that the previously reported associations between gut microbiota and health outcomes do not necessarily imply causality |
| Luo et al. (2023)[17] | Independent SNPS tightly associated with 196 gut bacterial taxa were used to ascertain the causal effect of intestinal microbiota on CKD with two-sample MR (n = 480,698) CKD was defined as eGFR < 60 mL/min/1.73m2 | The genetically predicted higher abundance of Desulfovibrionales was causally associated with higher odds of CKD Additionally, potentially significant causalities between nine other taxa and CKD were also identified | This study confirms that the intestinal microbiome is a major player in the pathogenesis of CKD |
As shown in Table 6, with one exception[108], studies consistently agree in supporting a causal association between gut microbiota and CKD. However, investigations fail to identify a unique “microbiological signature” that is associated with CKD and, probably, “dysbiosis”, namely the reduction in physiological diversity of intestinal microbiota, which is the shared common factor predisposing to the development of CKD. Dysbiosis leads to the so-called “leaky gut syndrome” or “endotoxemia”, which abrogates the intestine’s normal filter capacity and permits the passage of lipopolysaccharides and toxins of intestinal origin into the bloodstream, which may promote CKD via systemic inflammation, oxidative stress, and immune dysregulation[17,109]. To complicate things further, a bidirectional relationship links gut dysbiosis and CKD, and, in turn, CKD can lead to perturbed intestinal microecology[110].
In addition to the loss of functional integrity of the gut barrier, gut microbiota may chronically damage the kidneys either via the increased production of nephrotoxins, or through reduced production of beneficial substances that prevent nephrotoxicity.
Among the various nephrotoxins of intestinal origin, there are p-Cresol (p-C), Indoxyl Sulfate (IS), and p-Cresyl Sulfate (p-CS), which result from the fermentative activity of gut bacteria[111]. The blood levels of these compounds tend to increase among CKD patients in proportion to the severity of decreased GFR, since these metabolites are normally eliminated via the urinary route[111]. Collectively, P-CS, IS, and p-C, by activating chronic systemic inflammation, increasing the production of free radicals, and promoting immune dysfunction[111], may potentially promote the worsening of CKD and the development of CKD complications.
Trimethylamine N-oxide (TMAO) is a liver-synthesized compound [(synthesized from trimethylamine (TMA)], which derives from animal-derived choline and carnitine-rich foods by the action of the gut microbiota, and is finally excreted via renal route into the urine[112]. This explains why TMAO concentrations, compared to controls without CKD, are elevated in ESKD and hemodialysis patients[113]. Studies have disclosed the role of TMAO in cardiometabolic disorders including diabetes, HTN, cardiovascular disease, heart failure, and atrial fibrillation[114].
To provide a more comprehensive understanding of the relation between circulating TMAO concentrations and renal function, Zeng et al., in their meta-analytic review comprising 32 original publications totaling 42,062 individuals, found that circulating TMAO concentrations and renal function were inversely associated[113]. In detail, advanced CKD was associated with a 67.9 μmol/L increase in TMAO concentration, and significantly positively associated with various parameters assessing CKD severity[113].
Based on an analysis of 521 stable CKD subjects with CKD (defined with eGFR < 60 mL/min/1.73m2), followed for 5 years, Tang et al. found that plasma TMAO levels are elevated in patients with CKD and portend poorer long-term survival, suggesting that chronic dietary exposures that increase TMAO may contribute to renal fibrosis and progressive kidney dysfunction[115].
Short-chain fatty acids (SCFAs), including acetate, propionate, and butyrate, are saturated fatty acids with
Physiologically, butyrate represents a major energetic source for the intestinal epithelium through phosphorylation of AMPK and a stimulus for the release of glucagon-like peptide-1 (GLP1)[117]. After binding the G-protein-coupled receptor 41 (GPCR41) and activation of GPR41 in the intestinal epithelium, acetate and propionate promote the secretion of peptide YY (PYY) and control satiety and intestinal transit[117]. Additionally, GPR43 inhibits the production of proinflammatory factors and enhances GLP1 secretion, which induces the proliferation of pancreatic beta cells and thus exerts nephroprotection in DN by lowering glycemic levels[118].
Over the last few years, it has become increasingly clear that receptors of free fatty acids (FFAs) are a recently discovered class of GPCRs that account for agonist- and tissue-specific responses to dietary FFAs. In health, FFA receptor signaling promotes glucose-stimulated insulin secretion, homeostasis of enterohepatic cycle and enteroendocrine cells, and nutrient-sensitive energy regulation, and finally, it critically associates metabolic activities with immunological comebacks through regulation of inflammatory responses and secretion of peptide hormones[119,120]. The finding that GPR40 and GPR120 have been described in macrophages and neutrophils, respectively, two key cell types involved in the regulation of the innate immune response, raises the logical expectation that FFA receptor signaling may be leveraged to treat not only T2D but also NAFLD/NASH and related disorders[120,121].
Experimental evidence in STZ-induced diabetic mouse models shows that the gut dysbiosis-related low level of SCFAs in the intestinal tract in diabetic rodents is tightly associated with the initiation of DN[116]. Consistently, the administration of either SCFAs or GPR41 agonists can prevent incident DN through a variety of mechanisms: inhibited expansion of mesangial cells, reduced oxidative stress, and enhanced anti-inflammatory activity[122].
Recent investigation adds further evidence to the notion that SCFAs are causally associated with preserved kidney function. Mazidi et al. applied MR analysis to explore the relationships among genetically determined plasma valerate (an SCFA) with renal function and CKD risk[123]. While disclosing no significant association between plasma valerate and CKD, this study found plasma valerate to be directly associated with eGFR both in the overall population and among nondiabetic subjects. This investigation suggests the opportunity to conduct further research to clarify the links between plasma valerate, eGFR, and diabetes.
Portal hypertension
The finding that fibrosis is a major risk factor for incident CKD (summarized in Table 3) raises the logical expectation that portal hypertension might be mechanistically associated with deteriorated renal function. This hypothesis is strongly supported by a robust line of research indicating that, in rat models, fatty droplets within hepatocytes (and hepatocyte ballooning in NASH) distort the lumen of hepatic sinusoids and reduce it by up to 50%, therefore determining portal hypertension irrespective of fibrosis[124,125]. Francque et al., in their pioneering study, found elevated hepatic venous pressure gradient in 28% of 50 consecutive patients. In comparing those with and those without portal hypertension, the severity of steatosis was the only statistically significant histological parameter distinguishing between the two groups and predicted portal hypertension at regression analysis. Both parameters of visceral adiposity and IR were significantly associated with the presence of portal hypertension among those with severe steatosis[126]. More recently, this line of research has been developed further as discussed elsewhere by Lonardo et al.[43]. However, the mechanism(s) potentially conducive from uncomplicated portal hypertension to CKD remain(s) to be elucidated.
Figure 1 schematically recapitulates the most important factors contributing to CKD because of NAFLD/MAFLD/MASLD.
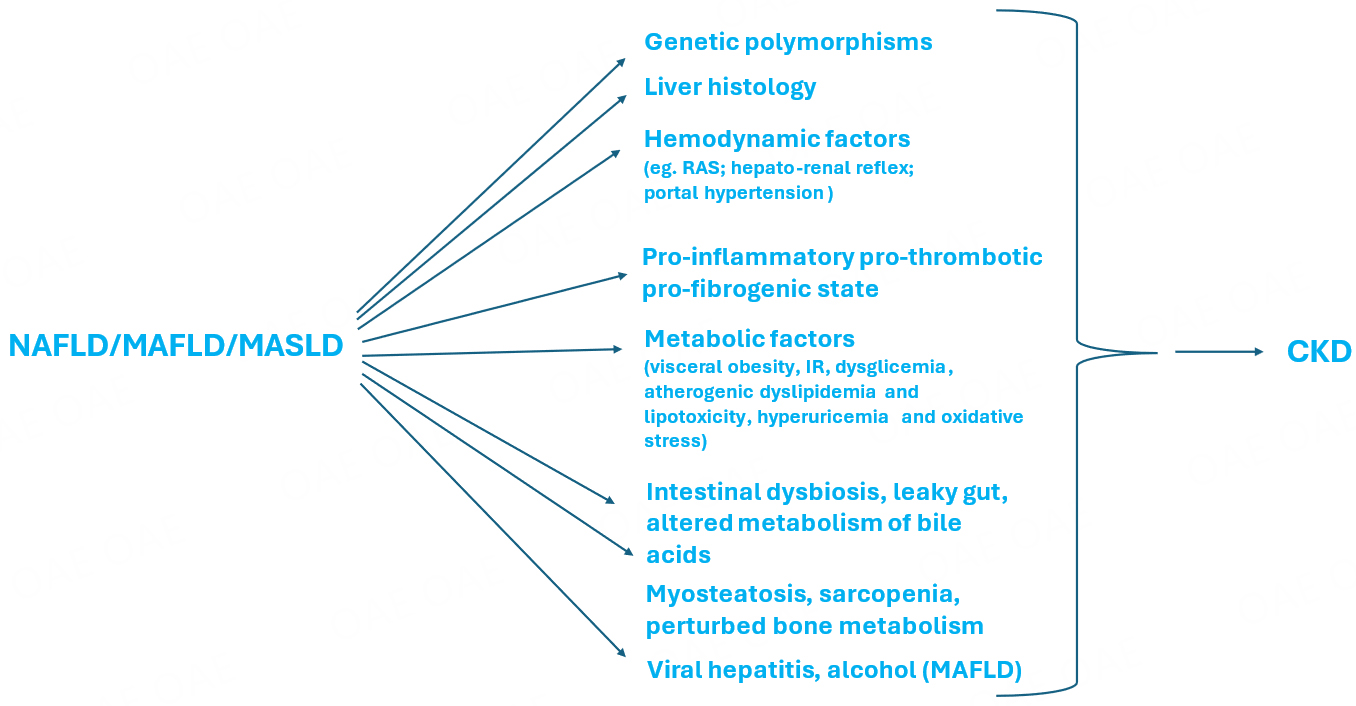
Figure 1. As analytically discussed in Chapter 3., genetic, liver-related, hemodynamic, metabolic, musculo-skeletal, and intestinal factors may mediate the development of incident CKD among those with NAFLD/MAFLD/MASLD. Additionally, viral hepatitis and alcohol may confer an additional risk of nephrotoxicity among a subset of individuals. CKD: chronic kidney disease; NAFLD: nonalcoholic fatty liver disease; MAFLD: metabolic dysfunction-associated fatty liver disease; MASLD: metabolic dysfunction-associated steatotic liver disease.
PREVENTION AND MANAGEMENT OF CKD AMONG THOSE WITH NAFLD/MAFLD/MASLD
Lifestyle changes comprising diet and exercise are the established mainstay of CKD prevention among those with NAFLD/MAFLD/MASLD. Pharmacotherapy for this population, on the other hand, appears to be in its early stages of development.
Attesting to the intimate relationship between improved liver histology and lifestyle changes, a recent investigation including 261 individuals with histologically-diagnosed NASH demonstrated that a one-stage reduction in liver fibrosis and NASH resolution was associated with improved parameters of renal function[127].
A study conducted among 3,926 participants found that, compared to those enrollees engaging self-reported moderate-to-vigorous physical activity classified within the lowest quartile, those individuals positioning themselves in the highest quartile had a reduced odds of atherosclerotic events, incident heart failure, and, importantly, all-cause and cardiovascular mortality[128]. More specifically, a large two-prospective cohort study from China found that, after 1,135,334 person-year follow-up among MAFLD patients, a healthier lifestyle was associated with a significantly reduced risk of CKD[57].
Dieting and direct manipulation of gut microbiota
Both under experimental conditions and in humans, unhealthy hypercaloric diets (featuring a high content of fructose and animal fat) may eventually lead to CKD via lipotoxicity resulting from the ectopic accumulation of fatty substrates in the peripheral organs (including the liver and kidneys), wherein metabolic inflammation, oxidative stress, fibrosis, and functional impairment eventually develop[10,129-132]. It is, therefore, logical to assume that reduced intake of energy, by restoring the body's ability to accumulate fat in the adipose tissue (as opposed to extra-adipose organs), will improve the imbalance of metabolic homeostasis and reverse the distribution of ectopic fat in the peripheral organs. However, lessons from extreme human phenotypes such as lipodystrophy[133] and clinical studies[134,135] disclose that, when it comes to fat, quality matters more than quantity.
Although substantial lifestyle changes have been advocated to treat CKD, diet remains relatively underused in the clinics[132,136]. Mechanistically, dietary manipulations might improve renal health via improved function and composition of gut microbiota and therefore changes in the spectrum of microbiota-derived metabolites that may be either nephroprotective (e.g., short-chain fatty acids) or detrimental to renal health (e.g., gut-derived uremic toxins)[136]. Intermittent fasting, a promising approach to delay the progression of CKD, remains under active investigation, particularly in the DN arena[137]. However, more robust evidence supports the notion that calorie restriction is beneficial for both NASH and kidney health[138,139].
Further to diets, additional approaches aimed at improving the composition of gut by correcting dysbiosis and restoring “eubiosis” include the supplementation of prebiotic, probiotic, and symbiotic principles, treatment of constipation, fecal microbiota transplantation, and intestinal dialysis[140]. Supplementation of polyphenol-rich berry fruits is associated with enhanced expression of mRNA of those proteins that are involved in preserving the function of intestinal tight junctions [i.e., occludin, tight junction protein 1 (TJP1), and mucin][141]. Therefore, clinical studies assessing the amounts and safety of wild berries are necessary to reduce toxin production, systemic inflammation, oxidative stress, and risk of cardiovascular disease, thus improving renal disease, quality of life, and prolonging the survival of subjects with CKD[141].
Exercise
Experimental evidence in mice suggests that endurance exercise training, via activation of the AMPK pathway in the renal tissue, improves various physiopathological aspects of ORN[142].
A nationwide Korean cohort study enrolling 7,275 participants from one cohort, and 40,418 participants with NAFLD from another cohort followed for a median 5.0-year time found that physical exercise was associated with a significantly reduced risk of CKD in subjects with NAFLD[143].
Lifestyle changes
Various items describe a “healthy lifestyle” including alcohol consumption, smoking, consumption of vegetables, avoidance of processed foodstuffs, and engaging in physical activity. It is reasonable to assume that the closer an individual’s lifestyle adheres to this healthy pattern, the more he/she is protected from incident CKD in the context of NAFLD/MAFLD/MASLD. Zhang et al. tested this hypothesis in two large prospective cohorts: the Chinese TCLSIH cohort including 25,974 participants, and the UK Biobank Study (UKB) comprising 113,954 participants[57]. CKD was defined by eGFR < 60 mL/min/1.73 m2, proteinuria, or a clinical diagnosis of CKD. The scores of the four established lifestyle habits predisposing to CKD, including smoking, alcohol consumption, physical activity, and dietary intake, were utilized for computing a healthy lifestyle score ranging from 0 to 4, such that the higher the score, the healthier the lifestyle. Finally, based on 263 single nucleotide polymorphisms (SNPs) that were associated with eGFR, a weighted GRS for eGFR was constructed for each participant. Data have shown that, after 1,135,334 person-year follow-up, adherence to ≥ 3 items of the healthy lifestyle score was associated with reduced risks of incident CKD among MAFLD patients[57]. A recent study including 17,040 participants from the NHANES:1998-2018 demonstrated that moderate alcohol consumption offers protection against CKD in men (but not women) with NAFLD[144]. Collectively, studies strongly support the notion that attaining blood pressure and glycemic targets is the backbone of care in preventing CKD progression[145].
Drugs
Standard of care in CKD arena: statins and renin-angiotensin system inhibitors
Statins and renin-angiotensin-aldosterone system (RAAS) inhibitors are deemed to be the standard of care among those at risk of CKD progression, although they were found to be underutilized in a recent large population-based retrospective cohort study conducted in Canada[146]. Sodium-glucose cotransporter 2 inhibitors (SGLT2i) have more recently been added to these[147]. Of interest, studies also support the utility of each of the above drug classes in the context of NAFLD/MAFLD/MASLD[148-150]. These studies raise the expectation of “killing two birds with one stone”, a notion that has previously been applied to the NAFLD arena associated with cardiovascular disorders[151].
SGLT2i and Glucagon-like peptide 1 receptor agonists
T2D is a driving etiology of CKD globally and simultaneous transplantation of kidney and liver is dramatically increasing in the United States, owing to NASH-cirrhosis being often accompanied by ESKD[152,153]. An exhaustive discussion of those antidiabetic agents that may also benefit NASH is out of the scope of this review and this topic has been extensively covered elsewhere[154]. Several molecules belonging to the classes of the Glucagon-like peptide 1 receptor agonists (GLP-1RA), including liraglutide and semaglutide, as well as of the SGLT2i such as canagliflozin and empagliflozin, have proven beneficial in reducing the odds of adverse renal and cardiovascular outcomes[155]. Strong evidence demonstrates that SGLT2i and GLP-1RA significantly reduce the risk of both CKD and CVD by improving the compensation of glucose homeostasis. Moreover, data strongly encourage the combination of SGLT2i with nonsteroidal mineralocorticoid receptor antagonists as a strategy that magnifies these cardiovascular and renal outcomes[152]. Interestingly, a meta-analytic review, updated as of December 2020 and based on 21 trials with 170,930 participants globally, found that SGLT2i was superior to GLP-1RA in reducing hospitalization for heart failure and renal outcomes, particularly among elderly, white, and Asian individuals, those subjects with long-standing or decompensated diabetes, and established atherosclerotic CVD and those with longer durations of diabetes mellitus and worse glycemic control[156]. A more recent meta-analysis of 17 eligible randomized controlled trials pooling data from 109,892 participants with T2D found that GLP-1RAs and SGLT2i not only offer cardiovascular benefits but also exert a positive impact on mortality[157]. Finally, a meta-analysis of 12 trials globally comprising 90,865 patients estimated meta-numbers needed to treat of 85 for GLP-1RA and 104 for SGLT2i (at the overall 36-month median follow-up), suggesting that both classes of drugs, GLP-1RA and SGLT2i, exert moderate and similar absolute treatment benefits for the composite renal outcome[158].
Finerenone
Finerenone, a novel non-steroidal mineralocorticoid receptor antagonist, represents a welcome addition to the arsenal for safeguarding kidney and cardiovascular system. Indeed, the current standard of care in diabetic kidney disease, while focusing on the control of glycemia and blood pressure, neglects inflammation and fibrosis. In preclinical models, finerenone effectively inhibited inflammatory, fibrotic, oxidative, and hypertrophic processes by blocking sodium reabsorption mediated by mineralocorticoid receptors as well as overactivation of mineralocorticoid receptors[159]. The FIDELITY study, a pooled analysis of two previously published studies, FIDELIO-DKD and FIGARO-DKD, assigned > 6,500 individuals with CKD and T2D to receive either finerenone (10 or 20 mg once daily) or placebo, in addition to the maximum tolerated renin-angiotensin system inhibition. Over a 3-year median follow-up, compared to placebo, finerenone was associated with a reduced risk of meaningful cardiovascular and kidney outcomes across the spectrum of CKD among T2D individuals[160]. The accompanying editorial for this publication concluded that Finerenone now stands alongside angiotensin-converting enzyme (ACE) inhibitors, SGTL2i, and GLP-1RA as a major contributor to reducing the risk of cardiovascular and kidney complications in individuals with T2D and CKD[161].
Pemafibrate
Conventional fibrates, including bezafibrate and fenofibrate, are agonists of peroxisome proliferator-activated receptor-alpha (PPARα)[162]. Given that PPARα concentrations are markedly reduced in the renal tissue of CKD individuals, therapeutic activity of fibrates against CKD would be expected[163,164].
Fibrates are generally associated with modest increases in creatinine levels when treatment is initiated. However, these increases tend to stabilize throughout the course of treatment and are reversible once fibrate therapy is discontinued[165]. Although robust data on the safety of fibrates in CKD are lacking and their capacity to delay ESKD remains uncertain, recent analysis suggests that fibrates, when administered against dyslipidemia, reduce the progression of albuminuria, and facilitate its regression among individuals with/without diabetes[165]. With this background of uncertainty, pemafibrate represents an evolution compared to the pre-existing fibrates.
Pemafibrate, a novel selective PPARα modulator with mainly biliary excretion, which has been shown to be effective in improving inflammatory cytokines and renal fibrosis and function in a mouse model of unilateral ureteral obstruction-induced CKD[163]. Studies have consistently shown that, in correcting dyslipidemia, pemafibrate has a good profile of safety and efficacy among CKD patients[164,166].
A recent study conducted in 47,490 Japanese patients with CKD (median follow-up of 9.4 months) found that pemafibrate use (rather than bezafibrate or fenofibrate use) was associated with a strongly decreased risk of major adverse cardiovascular events among patients with CKD (OR 0.73; 95%CI: 0.528-0.997)[167]. These intra-class differences result from pemafibrate, compared to older fibrates, exhibiting increased power and selectivity of PPAR alpha, which may, therefore, have more pronounced lipid-lowering and anti-inflammatory effects (documented by a decrease in C-reactive protein serum levels), with fewer drug-drug interactions and side effects[167].
Vonafexor
FXR agonists may represent a potentially useful therapeutic strategy to halt the progression of early-stage kidney disease to CKD[168]. Initially licensed for primary biliary cholangitis (PBC), obeticholic acid (OCA) was first tested in the NASH arena in the landmark FLINT trial[169].
Vonafexor (EYP001a) is a second-generation, synthetic, non-steroidal, non-bile acid, highly selective FXR agonist with a good profile of safety and efficacy at oral doses of up to 500 mg QD[170]. Compared to OCA, vonafexor promises improved efficacy in liver histology with reduced side effects.
Ratziu et al. conducted a double-blind phase IIa study named “LIVIFY trial”, globally randomizing 120 enrollees[170]. Patients were randomized to receive either placebo or vonafexor (at variable doses from 100 twice daily to 400 mg QD) to assess drug safety run-in, pharmacokinetics, and pharmacodynamics. Data have shown that, from the baseline to week 12, following drug treatment, there was a significant reduction in least-square mean (SE) absolute change in the primary outcome, i.e., liver fat content (LFC). Vonafexor was also associated with improved secondary outcomes. Mild to moderate generalized pruritus was reported in a dose-dependent manner in 9.7% to 18.2% of participants receiving vonafexor (vs. 6.3% in the placebo arm)[170]. Compared to placebo, vonafexor administration was associated with significantly improved eGFR, suggesting possibly improved kidney function. However, enthusiasm is mitigated by the failure to assess albuminuria quantitatively. Indeed, elevated eGFR associated with increased albuminuria would also be compatible with glomerular hyperfiltration, which is a potential precursor to CKD[171], as opposed to decreased or stable albuminuria, which would instead indicate true potential benefit in preventing/slowing long-term CKD in this context. Moreover, vonafexor-associated weight loss could account for reduced synthesis of creatinine and thereby raised eGFR irrespective of renal function, although simultaneously decreased serum uric acid concentrations among those randomized to vonafexor suggest really improved renal function. Collectively, these hopes and uncertainties call for additional investigation.
CONCLUSION
NAFLD/MAFLD/MASLD may predispose to incident CKD [Table 4], and CKD is an independent risk factor for mortality among NAFLD patients with diabetes[172]. Moreover, patients with CKD and NAFLD exhibit a higher risk of CVE, and the NAFLD fibrosis score predicts an elevated risk of CVE and decreased life expectancy[173-175].
The association of baseline NAFLD with incident CKD is only one side of the coin as individuals in whom CKD at the baseline is associated with co-morbid conditions such as diabetes, obesity, cardiac disease, and anemia also face a heightened risk of incident NAFLD[176]. More broadly, the presence of CKD deeply affects the outcomes of patients with hepatic cirrhosis owing to the increased risks of acute kidney injury, need for dialytic treatment, acute-on-chronic liver failure, and decreased life expectancy at 30 days[177].
These observations strongly support the opportunity to prevent the development of CKD among those with NAFLD/MAFLD/MASLD. Available drugs affect the principal pathogenic pathways involved in the development of NAFLD/MAFLD/MASLD: cholesterol synthesis and nuclear receptors[178,179]. Additional investigations are needed to ascertain the role of innovative therapeutic strategies aimed at inhibiting renal interstitial fibrosis by blocking the epithelial-mesenchymal transition process[89].
CKD is closely associated with the MetS and is an integral part of the CKM[13,14].
Projections indicate that by 2040, CKD will rank as the fifth leading cause of mortality. This places CKD among the few non-transmissible diseases that are progressively claiming more lives, marking it as one of the fastest-growing causes of mortality over the last two decades[27]. These alarming figures underscore the urgency of efforts aimed at halting this silent CKD epidemic. Given the multiplicity of pathomechanisms involved, targeting NAFLD/MAFLD/MASLD could be a rational option to combat CKD, especially considering their strong association with incident CKD in a manner that parallels the severity of liver disease [Tables 3 and 4].
However, the spectrum of contributors to the initiation and worsening of incident CKD among those with NAFLD/MAFLD/MASLD is not yet fully understood and needs further examination. For example, the role of the dyad comprising skeletal muscle and bone in this arena needs further characterization. This expectation is based on the finding that myosteatosis and sarcopenia are involved in the severity of NAFLD and MAFLD[180,181] and that advanced CKD involves a process of pseudo-ossification of media of large- and medium-caliber vessels that strongly contributes to heart failure[182,183]. Finally, the role of the liver in the risk of incident CKD also needs to be further characterized with specific reference to portal hypertension[43].
Recently, the changing definitions of NAFLD/MAFLD/MASLD have resulted in confusion among physicians[5]. While the NAFLD spectrum describes a diagnosis of exclusion (i.e., “nonalcoholic”)[184], the MAFLD nomenclature identifies a positive diagnostic criterion (i.e., “metabolic dysfunction”). Metabolic dysfunction is an array of cardiovascular risk factors including visceral adiposity, arterial hypertension, hyperinsulinemia/insulin resistance, altered glucose metabolism, pro-atherogenic dyslipidemia, and low-grade subclinical inflammation[185], all of which carry increased odds of CKD, suggesting that metabolic dysfunction is a major mechanistic connector of MAFLD with CKD. Such a connection occurs via the secretion of the adipokines leptin and adiponectin, which dictate satiety, govern hepatic and systemic insulin sensitivity, low-grade chronic inflammation and the renin-angiotensin system, promote podocyte viability, govern morphogenesis of liver histology elementary changes (steatogenesis, hepatitis, and fibrogenesis), and contribute to the development and worsening of CKD by regulating renal hemodynamics via the sympathetic nervous system[38]. Another reason why MAFLD is superior to the NAFLD/NASH nomenclature is the notion that MAFLD (not NAFLD) can coexist with concurrent causes of chronic liver disease such as infections with major hepatitis viruses: Hepatitis B Virus (HBV) or Hepatitis C Virus (HCV) which may cause viral- related glomerulonephritides, suggesting that HBV and HCV could, in principle, account for the close association of MAFLD with CKD, although additional studies are necessary to address this point[38].
RESEARCH AGENDA
It has recently been anticipated that, by mid-2024, all human studies on NAFLD/MAFLD/MASLD will adhere to the new specified nomenclature and definitions[186]. However, specifically regarding the CKD arena, the potential benefits of these nomenclature changes remain uncertain given that the MAFLD definition probably “captures” the risk of incident CKD risk better than NAFLD in adults[50] but not in children[187]. Moreover, MASLD may inappropriately rule out patients with significant liver fibrosis, particularly lean women with NAFLD[188], and may otherwise probably overlap with NAFLD as far as the natural history is concerned[8]. In this connection, comparative studies between the various NAFLD/MAFLD/MASLD nomenclatures would be hampered if the adoption of the MASLD nosography were to be universally and abruptly adopted[189].
DECLARATIONS
Authors’ contributions
The author contributed solely to the article.
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
Amedeo Lonardo is the Editor-in-Chief of the journal Metabolism and Target Organ Damage.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2024.
REFERENCES
1. Lonardo A, Leoni S, Alswat KA, Fouad Y. History of nonalcoholic fatty liver disease. Int J Mol Sci 2020;21:5888.
2. Loria P, Lonardo A, Carulli N. Should nonalcoholic fatty liver disease be renamed? Dig Dis 2005;23:72-82.
3. Méndez-Sánchez N, Bugianesi E, Gish RG, et al. Global multi-stakeholder consensus on the redefinition of fatty liver disease. Global multi-stakeholder endorsement of the MAFLD definition. Lancet Gastroenterol Hepatol 2022;7:388-90.
4. Rinella ME, Lazarus JV, Ratziu V, et al. NAFLD Nomenclature consensus group. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. J Hepatol 2023;79:1542-56.
5. Bilson J, Mantovani A, Byrne CD, Targher G. Steatotic liver disease, MASLD and risk of chronic kidney disease. Diabetes Metab 2024;50:101506.
6. Byrne CD, Targher G. MASLD, MAFLD, or NAFLD criteria: have we re-created the confusion and acrimony surrounding metabolic syndrome? Metab Target Organ Damage 2024;4:10.
7. Duseja A, Singh SP, De A, et al. Indian national association for study of the liver (INASL) guidance paper on nomenclature, diagnosis and treatment of nonalcoholic fatty liver disease (NAFLD). J Clin Exp Hepatol 2023;13:273-302.
8. Kaya E, Yilmaz Y. Epidemiology, natural history, and diagnosis of metabolic dysfunction-associated fatty liver disease: a comparative review with nonalcoholic fatty liver disease. Ther Adv Endocrinol Metab 2022;13:20420188221139650.
9. Huang XJ, Yin M, Zhou BQ, Tan XY, Xia YQ, Qin CX. Impact renaming non-alcoholic fatty liver disease to metabolic associated fatty liver disease in prevalence, characteristics and risk factors. World J Hepatol 2023;15:985-1000.
10. Hagström H, Vessby J, Ekstedt M, Shang Y. 99% of patients with NAFLD meet MASLD criteria and natural history is therefore identical. J Hepatol 2024;80:e76-7.
11. Vaidya SR, Aeddula NR. Chronic renal failure. Available from: https://www.ncbi.nlm.nih.gov/books/NBK535404/ [Last accessed on 1 Apr 2024].
12. Ryu H, Hong Y, Kang E, et al. KNOW-CKD Study Group. Comparison of outcomes of chronic kidney disease based on etiology: a prospective cohort study from KNOW-CKD. Sci Rep 2023;13:3570.
13. Chen J, Muntner P, Hamm LL, et al. The metabolic syndrome and chronic kidney disease in U.S. adults. Ann Intern Med 2004;140:167-74.
14. Ndumele CE, Rangaswami J, Chow SL, et al. American Heart Association. Cardiovascular-kidney-metabolic health: a presidential advisory from the american heart association. Circulation 2023;148:1606-35.
15. Matsushita K, Ballew SH, Wang AY, Kalyesubula R, Schaeffner E, Agarwal R. Epidemiology and risk of cardiovascular disease in populations with chronic kidney disease. Nat Rev Nephrol 2022;18:696-707.
16. Zoccali C, Mallamaci F, Adamczak M, et al. Cardiovascular complications in chronic kidney disease: a review from the European renal and cardiovascular medicine working group of the European renal association. Cardiovasc Res 2023;119:2017-32.
17. Luo M, Cai J, Luo S, et al. Causal effects of gut microbiota on the risk of chronic kidney disease: a Mendelian randomization study. Front Cell Infect Microbiol 2023;13:1142140.
18. Riazi K, Azhari H, Charette JH, et al. The prevalence and incidence of NAFLD worldwide: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol 2022;7:851-61.
19. Quek J, Chan KE, Wong ZY, et al. Global prevalence of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in the overweight and obese population: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol 2023;8:20-30.
20. En Li Cho E, Ang CZ, Quek J, et al. Global prevalence of non-alcoholic fatty liver disease in type 2 diabetes mellitus: an updated systematic review and meta-analysis. Gut 2023;72:2138-48.
21. Ye Q, Zou B, Yeo YH, et al. Global prevalence, incidence, and outcomes of non-obese or lean non-alcoholic fatty liver disease: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol 2020;5:739-52.
22. Liu J, Tian Y, Fu X, et al. Estimating global prevalence, incidence, and outcomes of non-alcoholic fatty liver disease from 2000 to 2021: systematic review and meta-analysis. Chin Med J 2022;135:1682-91.
23. Xiao J, Ng CH, Chan KE, et al. Hepatic, extra-hepatic outcomes and causes of mortality in NAFLD - an umbrella overview of systematic review of meta-analysis. J Clin Exp Hepatol 2023;13:656-65.
24. Stepanova M, De Avila L, Afendy M, et al. Direct and indirect economic burden of chronic liver disease in the United States. Clin Gastroenterol Hepatol 2017;15:759-66.e5.
25. Sayiner M, Arshad T, Golabi P, Paik J, Farhat F, Younossi ZM. Extrahepatic manifestations and healthcare expenditures of non-alcoholic fatty liver disease in the Medicare population. Hepatol Int 2020;14:556-66.
26. Perazzo H, Pacheco AG, Griep RH. Collaborators. Changing from NAFLD through MAFLD to MASLD: similar prevalence and risk factors in a large Brazilian cohort. J Hepatol 2024;80:e72-4.
27. Kovesdy CP. Epidemiology of chronic kidney disease: an update 2022. Kidney Int Suppl 2022;12:7-11.
28. Arabi T, Shafqat A, Sabbah BN, et al. Obesity-related kidney disease: Beyond hypertension and insulin-resistance. Front Endocrinol 2022;13:1095211.
29. Zheng J, Zhang Y, Rasheed H, et al. Trans-ethnic Mendelian-randomization study reveals causal relationships between cardiometabolic factors and chronic kidney disease. Int J Epidemiol 2022;50:1995-2010.
30. Tsao HM, Lai TS, Chang YC, et al. Serum urate and risk of chronic kidney disease: a mendelian randomization study using Taiwan biobank. Mayo Clin Proc 2023;98:513-21.
31. Deprince A, Haas JT, Staels B. Dysregulated lipid metabolism links NAFLD to cardiovascular disease. Mol Metab 2020;42:101092.
32. Kim S, Chang Y, Sung E, et al. Non-alcoholic fatty liver disease and the development of nephrolithiasis: A cohort study. PLoS One 2017;12:e0184506.
33. Liu Z, Wang Q, Huang H, Wang X, Xu C. Association between serum uric acid levels and long-term mortality of metabolic dysfunction-associated fatty liver disease: a nationwide cohort study. Diabetol Metab Syndr 2023;15:27.
34. Chen TK, Knicely DH, Grams ME. Chronic kidney disease diagnosis and management: a review. JAMA 2019;322:1294-304.
35. Kalantar-Zadeh K, Jafar TH, Nitsch D, Neuen BL, Perkovic V. Chronic kidney disease. Lancet 2021;398:786-802.
36. Sanchez-Niño MD, Sanz AB, Ramos AM, Ruiz-Ortega M, Ortiz A. Translational science in chronic kidney disease. Clin Sci 2017;131:1617-29.
37. Gandjour A, Armsen W, Wehmeyer W, Multmeier J, Tschulena U. Costs of patients with chronic kidney disease in Germany. PLoS One 2020;15:e0231375.
38. Wang TY, Wang RF, Bu ZY, et al. Association of metabolic dysfunction-associated fatty liver disease with kidney disease. Nat Rev Nephrol 2022;18:259-68.
39. Mantovani A, Lombardi R, Cattazzo F, Zusi C, Cappelli D, Dalbeni A. MAFLD and CKD: an updated narrative review. Int J Mol Sci 2022;23:7007.
40. Theofilis P, Vordoni A, Kalaitzidis RG. Interplay between metabolic dysfunction-associated fatty liver disease and chronic kidney disease: Epidemiology, pathophysiologic mechanisms, and treatment considerations. World J Gastroenterol 2022;28:5691-706.
41. Sun DQ, Targher G, Byrne CD, et al. An international Delphi consensus statement on metabolic dysfunction-associated fatty liver disease and risk of chronic kidney disease. Hepatobiliary Surg Nutr 2023;12:386-403.
42. Nysather J, Kaya E, Manka P, Gudsoorkar P, Syn WK. Nonalcoholic fatty liver disease and chronic kidney disease cross talk. Adv Kidney Dis Health 2023;30:315-35.
43. Lonardo A, Mantovani A, Targher G, Baffy G. Nonalcoholic fatty liver disease and chronic kidney disease: epidemiology, pathogenesis, and clinical and research implications. Int J Mol Sci 2022;23:13320.
44. Musso G, Gambino R, Tabibian JH, et al. Association of non-alcoholic fatty liver disease with chronic kidney disease: a systematic review and meta-analysis. PLoS Med 2014;11:e1001680.
45. Liu HW, Liu JS, Kuo KL. Association of nonalcoholic fatty liver and chronic kidney disease: An analysis of 37,825 cases from health checkup center in Taiwan. Ci Ji Yi Xue Za Zhi 2020;32:65-9.
46. Akahane T, Akahane M, Namisaki T, et al. Association between non-alcoholic fatty liver disease and chronic kidney disease: a cross-sectional study. J Clin Med 2020;9:1635.
47. Deng Y, Zhao Q, Gong R. Association between metabolic associated fatty liver disease and chronic kidney disease: a cross-sectional study from NHANES 2017-2018. Diabetes Metab Syndr Obes 2021;14:1751-61.
49. Su W, Chen M, Xiao L, et al. Association of metabolic dysfunction-associated fatty liver disease, type 2 diabetes mellitus, and metabolic goal achievement with risk of chronic kidney disease. Front Public Health 2022;10:1047794.
50. Agustanti N, Soetedjo NNM, Damara FA, et al. The association between metabolic dysfunction-associated fatty liver disease and chronic kidney disease: a systematic review and meta-analysis. Diabetes Metab Syndr 2023;17:102780.
51. Mantovani A, Petracca G, Beatrice G, et al. Non-alcoholic fatty liver disease and risk of incident chronic kidney disease: an updated meta-analysis. Gut 2022;71:156-62.
52. Mantovani A, Petracca G, Beatrice G, et al. Non-alcoholic fatty liver disease and risk of incident chronic kidney disease: an updated meta-analysis. Gut 2022;71:156-62.
53. Yi M, Peng W, Feng X, et al. Extrahepatic morbidities and mortality of NAFLD: an umbrella review of meta-analyses. Aliment Pharmacol Ther 2022;56:1119-30.
54. Tanaka M, Mori K, Takahashi S, et al. Metabolic dysfunction-associated fatty liver disease predicts new onset of chronic kidney disease better than fatty liver or nonalcoholic fatty liver disease. Nephrol Dial Transplant 2023;38:700-11.
55. Jung CY, Koh HB, Park KH, et al. Metabolic dysfunction-associated fatty liver disease and risk of incident chronic kidney disease: A nationwide cohort study. Diabetes Metab 2022;48:101344.
56. Hashimoto Y, Hamaguchi M, Okamura T, et al. Metabolic associated fatty liver disease is a risk factor for chronic kidney disease. J Diabetes Investig 2022;13:308-16.
57. Zhang Y, Zhang T, Liu Y, et al. Adherence to healthy lifestyle was associated with an attenuation of the risk of chronic kidney disease from metabolic dysfunction-associated fatty liver disease: results from two prospective cohorts. Diabetes Metab Syndr 2023;17:102873.
58. Pan LL, Zhang HJ, Huang ZF, et al. Intrahepatic triglyceride content is independently associated with chronic kidney disease in obese adults: a cross-sectional study. Metabolism 2015;64:1077-85.
59. Zuo G, Xuan L, Xin Z, et al. New nonalcoholic fatty liver disease and fibrosis progression associate with the risk of incident chronic kidney disease. J Clin Endocrinol Metab 2021;106:e3957-68.
60. Ciardullo S, Ballabeni C, Trevisan R, Perseghin G. Liver stiffness, albuminuria and chronic kidney disease in patients with NAFLD: a systematic review and meta-analysis. Biomolecules 2022;12:105.
61. Seo DH, Suh YJ, Cho Y, et al. Advanced liver fibrosis is associated with chronic kidney disease in patients with type 2 diabetes mellitus and nonalcoholic fatty liver disease. Diabetes Metab J 2022;46:630-9.
62. Sun Y, Hong L, Huang Z, et al. Fibrosis risk in nonalcoholic fatty liver disease is related to chronic kidney disease in older type 2 diabetes patients. J Clin Endocrinol Metab 2022;107:e3661-9.
63. Chung GE, Han K, Lee KN, et al. Association between fatty liver index and risk of end-stage renal disease stratified by kidney function in patients with type 2 diabetes: a nationwide population-based study. Diabetes Metab 2023;49:101454.
64. Lonardo A. The heterogeneity of metabolic syndrome presentation and challenges this causes in its pharmacological management: a narrative review focusing on principal risk modifiers. Expert Rev Clin Pharmacol 2023;16:891-911.
65. Machado MV, Michelotti GA, Xie G, et al. Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS One 2015;10:e0127991.
67. Wentworth BJ, Caldwell SH. Pearls and pitfalls in nonalcoholic fatty liver disease: tricky results are common. Metab Target Organ Damage 2021;1:2.
68. Arrese M, Arab JP, Barrera F, Kaufmann B, Valenti L, Feldstein AE. Insights into nonalcoholic fatty-liver disease heterogeneity. Semin Liver Dis 2021;41:421-34.
69. Pal P, Palui R, Ray S. Heterogeneity of non-alcoholic fatty liver disease: implications for clinical practice and research activity. World J Hepatol 2021;13:1584-610.
70. Lonardo A, Singal AK, Osna N, Kharbanda KK. Effect of cofactors on NAFLD/NASH and MAFLD - a paradigm illustrating the pathomechanics of organ dysfunction. Metab Target Organ Damage 2022;2:12.
71. Baratta F, D'Erasmo L, Bini S, et al. Heterogeneity of non-alcoholic fatty liver disease (NAFLD): implication for cardiovascular risk stratification. Atherosclerosis 2022;357:51-9.
72. Pirola CJ, Sookoian S. Advances in our understanding of the molecular heterogeneity of fatty liver disease: toward informed treatment decision making. Expert Rev Gastroenterol Hepatol 2023;17:317-24.
73. Trépo E, Valenti L. Update on NAFLD genetics: From new variants to the clinic. J Hepatol 2020;72:1196-209.
74. Lonardo A. Principles of risk stratification in nonalcoholic fatty liver disease. A narrative review emphasizing non-invasive strategies. Explor Dig Dis ;2:188-201.
75. Sun DQ, Zheng KI, Xu G, et al. PNPLA3 rs738409 is associated with renal glomerular and tubular injury in NAFLD patients with persistently normal ALT levels. Liver Int 2020;40:107-19.
76. Mantovani A, Taliento A, Zusi C, et al. PNPLA3 I148M gene variant and chronic kidney disease in type 2 diabetic patients with NAFLD: clinical and experimental findings. Liver Int 2020;40:1130-41.
77. Akuta N, Kawamura Y, Arase Y, et al. PNPLA3 genotype and fibrosis-4 index predict cardiovascular diseases of Japanese patients with histopathologically-confirmed NAFLD. BMC Gastroenterol 2021;21:434.
78. Mantovani A, Pelusi S, Margarita S, et al. Adverse effect of PNPLA3 p.I148M genetic variant on kidney function in middle-aged individuals with metabolic dysfunction. Aliment Pharmacol Ther 2023;57:1093-102.
79. Mantovani A, Targher G. PNPLA3 rs738409 polymorphism and kidney dysfunction: an association beyond nonalcoholic fatty liver disease? Metab Target Organ Damage 2023;3:18.
80. Lonardo A, Ballestri S, Targher G. "Not all forms of NAFLD were created equal". Do metabolic syndrome-related NAFLD and PNPLA3-related NAFLD exert a variable impact on the risk of early carotid atherosclerosis? Atherosclerosis 2017;257:253-5.
81. Nashar K, Egan BM. Relationship between chronic kidney disease and metabolic syndrome: current perspectives. Diabetes Metab Syndr Obes 2014;7:421-35.
82. Selby NM, Taal MW. An updated overview of diabetic nephropathy: diagnosis, prognosis, treatment goals and latest guidelines. Diabetes Obes Metab 2020;22 Suppl 1:3-15.
83. Iqbal J, Wu HX, Nawaz MA, et al. Risk of incident chronic kidney disease in metabolically healthy obesity and metabolically unhealthy normal weight: a systematic review and meta-analysis. Obes Rev 2024;25:e13656.
85. Masenga SK, Kabwe LS, Chakulya M, Kirabo A. Mechanisms of oxidative stress in metabolic syndrome. Int J Mol Sci 2023;24:7898.
86. Stenvinkel P, Chertow GM, Devarajan P, et al. Chronic inflammation in chronic kidney disease progression: role of Nrf2. Kidney Int Rep 2021;6:1775-87.
87. Dorotea D, Koya D, Ha H. Recent insights into SREBP as a direct mediator of kidney fibrosis via lipid-independent pathways. Front Pharmacol 2020;11:265.
88. Avraham S, Korin B, Chung JJ, Oxburgh L, Shaw AS. The mesangial cell - the glomerular stromal cell. Nat Rev Nephrol 2021;17:855-64.
89. Costantino VV, Gil Lorenzo AF, Bocanegra V, Vallés PG. Molecular mechanisms of hypertensive nephropathy: renoprotective effect of losartan through Hsp70. Cells 2021;10:3146.
90. Eshraghi Y, Abedi M, Gheisari Y. Proteomics to metabolomics: a new insight into the pathogenesis of hypertensive nephropathy. Kidney Blood Press Res 2023;48:710-26.
91. Alicic RZ, Rooney MT, Tuttle KR. Diabetic kidney disease: challenges, progress, and possibilities. Clin J Am Soc Nephrol 2017;12:2032-45.
92. DeFronzo RA, Reeves WB, Awad AS. Pathophysiology of diabetic kidney disease: impact of SGLT2 inhibitors. Nat Rev Nephrol 2021;17:319-34.
93. Berfield AK, Andress DL, Abrass CK. IGF-1-induced lipid accumulation impairs mesangial cell migration and contractile function. Kidney Int 2002;62:1229-37.
94. Guo Y, Xie G, Zhang X. Role of FXR in renal physiology and kidney diseases. Int J Mol Sci 2023;24:2408.
95. Zhu JB, Xu S, Li J, et al. Farnesoid X receptor agonist obeticholic acid inhibits renal inflammation and oxidative stress during lipopolysaccharide-induced acute kidney injury. Eur J Pharmacol 2018;838:60-8.
96. Palladini G, Cagna M, Di Pasqua LG, et al. Obeticholic acid reduces kidney matrix metalloproteinase activation following partial hepatic ischemia/reperfusion injury in rats. Pharmaceuticals 2022;15:524.
97. Gege C, Hambruch E, Hambruch N, Kinzel O, Kremoser C. Nonsteroidal FXR ligands: current status and clinical applications. In: Fiorucci S, Distrutti E, editors. Bile Acids and Their Receptors. Cham: Springer International Publishing; 2019. pp. 167-205.
98. Andres-Hernando A, Lanaspa MA, Kuwabara M, et al. Obesity causes renal mitochondrial dysfunction and energy imbalance and accelerates chronic kidney disease in mice. Am J Physiol Renal Physiol 2019;317:F941-8.
99. Søgaard SB, Andersen SB, Taghavi I, et al. Super-resolution ultrasound imaging provides quantification of the renal cortical and medullary vasculature in obese zucker rats: a pilot study. Diagnostics 2022;12:1626.
100. Hashemi L, Hsiung JT, Arif Y, et al. Serum low-density lipoprotein cholesterol and cardiovascular disease risk across chronic kidney disease stages (data from 1.9 million United States veterans). Am J Cardiol 2022;170:47-55.
101. Baragetti A, Norata GD, Sarcina C, et al. High density lipoprotein cholesterol levels are an independent predictor of the progression of chronic kidney disease. J Intern Med 2013;274:252-62.
102. Baragetti A, Ossoli A, Strazzella A, et al. Low plasma lecithin: cholesterol acyltransferase (LCAT) concentration predicts chronic kidney disease. J Clin Med 2020;9:2289.
103. Guerra S, Mocciaro G, Gastaldelli A. Adipose tissue insulin resistance and lipidome alterations as the characterizing factors of non-alcoholic steatohepatitis. Eur J Clin Invest 2022;52:e13695.
104. Jia J, Dou P, Gao M, et al. Assessment of causal direction between gut microbiota-dependent metabolites and cardiometabolic health: a bidirectional mendelian randomization analysis. Diabetes 2019;68:1747-55.
105. Mazidi M, Shekoohi N, Covic A, Mikhailidis DP, Banach M. Adverse impact of desulfovibrio spp. and beneficial role of anaerostipes spp. on renal function: insights from a mendelian randomization analysis. Nutrients 2020;12:2216.
106. Luo Q, Hu Y, Chen X, Luo Y, Chen J, Wang H. Effects of gut microbiota and metabolites on heart failure and its risk factors: a two-sample mendelian randomization study. Front Nutr 2022;9:899746.
107. Li N, Wang Y, Wei P, et al. Causal effects of specific gut microbiota on chronic kidney diseases and renal function-a two-sample mendelian randomization study. Nutrients 2023;15:360.
108. Gagnon E, Mitchell PL, Manikpurage HD, et al. Impact of the gut microbiota and associated metabolites on cardiometabolic traits, chronic diseases and human longevity: a Mendelian randomization study. J Transl Med 2023;21:60.
109. Pantazi AC, Kassim MAK, Nori W, et al. Clinical perspectives of gut microbiota in patients with chronic kidney disease and end-stage kidney disease: where do we stand? Biomedicines 2023;11:2480.
110. Feng Z, Wang T, Dong S, et al. Association between gut dysbiosis and chronic kidney disease: a narrative review of the literature. J Int Med Res 2021;49:3000605211053276.
111. Di Paola R, De A, Izhar R, et al. Possible effects of uremic toxins p-cresol, indoxyl sulfate, p-cresyl sulfate on the development and progression of colon cancer in patients with chronic renal failure. Genes 2023;14:1257.
112. Velasquez MT, Ramezani A, Manal A, Raj DS. Trimethylamine N-Oxide: the good, the bad and the unknown. Toxins 2016;8:326.
113. Zeng Y, Guo M, Fang X, et al. Gut microbiota-derived trimethylamine N-oxide and kidney function: a systematic review and meta-analysis. Adv Nutr 2021;12:1286-304.
114. Papandreou C, Moré M, Bellamine A. Trimethylamine N-oxide in relation to cardiometabolic health-cause or effect? Nutrients 2020;12:1330.
115. Tang WH, Wang Z, Kennedy DJ, et al. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res 2015;116:448-55.
116. Tao P, Ji J, Wang Q, Cui M, Cao M, Xu Y. The role and mechanism of gut microbiota-derived short-chain fatty in the prevention and treatment of diabetic kidney disease. Front Immunol 2022;13:1080456.
117. Portincasa P, Bonfrate L, Vacca M, et al. Gut microbiota and short chain fatty acids: implications in glucose homeostasis. Int J Mol Sci 2022;23:1105.
119. Kimura I, Ichimura A, Ohue-Kitano R, Igarashi M. Free fatty acid receptors in health and disease. Physiol Rev 2020;100:171-210.
120. Secor JD, Fligor SC, Tsikis ST, Yu LJ, Puder M. Free fatty acid receptors as mediators and therapeutic targets in liver disease. Front Physiol 2021;12:656441.
121. Hidalgo MA, Carretta MD, Burgos RA. Long chain fatty acids as modulators of immune cells function: contribution of FFA1 and FFA4 receptors. Front Physiol 2021;12:668330.
122. Kim MH, Kang SG, Park JH, Yanagisawa M, Kim CH. Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology 2013;145:396-406.e1.
123. Mazidi M, Katsiki N, Banach M. Higher plasma levels of valerate produced by gut microbiota may have a beneficial impact on renal function. J Am Nutr Assoc 2023;42:534-40.
124. Farrell GC, Teoh NC, McCuskey RS. Hepatic microcirculation in fatty liver disease. Anat Rec 2008;291:684-92.
125. Francque S, Wamutu S, Chatterjee S, et al. Non-alcoholic steatohepatitis induces non-fibrosis-related portal hypertension associated with splanchnic vasodilation and signs of a hyperdynamic circulation in vitro and in vivo in a rat model. Liver Int 2010;30:365-75.
126. Francque S, Verrijken A, Mertens I, et al. Visceral adiposity and insulin resistance are independent predictors of the presence of non-cirrhotic NAFLD-related portal hypertension. Int J Obes 2011;35:270-8.
127. Vilar-Gomez E, Calzadilla-Bertot L, Friedman SL, et al. Improvement in liver histology due to lifestyle modification is independently associated with improved kidney function in patients with non-alcoholic steatohepatitis. Aliment Pharmacol Ther 2017;45:332-44.
128. Bruinius JW, Hannan M, Chen J, et al. CRIC Study Investigators. Self-reported physical activity and cardiovascular events in adults with CKD: findings from the CRIC (chronic renal insufficiency cohort) study. Am J Kidney Dis 2022;80:751-61.e1.
129. Hamada S, Takata T, Yamada K, et al. Steatosis is involved in the progression of kidney disease in a high-fat-diet-induced non-alcoholic steatohepatitis mouse model. PLoS One 2022;17:e0265461.
130. Bier A, Shapira E, Khasbab R, Sharabi Y, Grossman E, Leibowitz A. High-fructose diet increases renal ChREBPβ expression, leading to intrarenal fat accumulation in a rat model with metabolic syndrome. Biology 2022;11:618.
131. Abbate M, Mascaró CM, Montemayor S, et al. Animal fat intake is associated with albuminuria in patients with non-alcoholic fatty liver disease and metabolic syndrome. Nutrients 2021;13:1548.
132. Meléndez-Salcido CG, Ramírez-Emiliano J, Pérez-Vázquez V. Hypercaloric diet promotes metabolic disorders and impaired kidney function. Curr Pharm Des 2022;28:3127-39.
133. Mann JP, Savage DB. What lipodystrophies teach us about the metabolic syndrome. J Clin Invest 2019;129:4009-21.
134. Kalavalapalli S, Leiva EG, Lomonaco R, et al. Adipose tissue insulin resistance predicts the severity of liver fibrosis in patients with type 2 diabetes and NAFLD. J Clin Endocrinol Metab 2023;108:1192-201.
135. Mocciaro G, Gastaldelli A. Obesity-related insulin resistance: the central role of adipose tissue dysfunction. In: Eckel J, Clément K, editors. From Obesity to Diabetes. Cham: Springer International Publishing; 2022. pp. 145-64.
136. Koppe L, Soulage CO. The impact of dietary nutrient intake on gut microbiota in the progression and complications of chronic kidney disease. Kidney Int 2022;102:728-39.
137. Yang M, Chen W, He L, Liu D, Zhao L, Wang X. Intermittent fasting-a healthy dietary pattern for diabetic nephropathy. Nutrients 2022;14:3995.
138. Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology 2015;149:367-78.e5; quiz e14.
139. Ruggenenti P, Abbate M, Ruggiero B, et al. CRE.S.O. Study Group. . Renal and systemic effects of calorie restriction in patients with type 2 diabetes with abdominal obesity: a randomized controlled trial. Diabetes 2017;66:75-86.
140. Sumida K, Pierre JF, Yuzefpolskaya M, Colombo PC, Demmer RT, Kovesdy CP. Gut microbiota-targeted interventions in the management of chronic kidney disease. Semin Nephrol 2023;43:151408.
141. Coutinho-Wolino KS, Melo MFS, Mota JC, Mafra D, Guimarães JT, Stockler-Pinto MB. Blueberry, cranberry, raspberry, and strawberry as modulators of the gut microbiota: target for treatment of gut dysbiosis in chronic kidney disease? Nutr Rev 2024;82:248-61.
142. Juszczak F, Vlassembrouck M, Botton O, et al. Delayed exercise training improves obesity-induced chronic kidney disease by activating AMPK pathway in high-fat diet-fed mice. Int J Mol Sci 2020;22:350.
143. Jung CY, Chun HS, Lee M, et al. Exercise reduces the risk of chronic kidney disease in individuals with nonalcoholic fatty liver disease: a nationwide cohort study. Diabetes Metab 2022;48:101362.
144. Zheng T, Wang X, Kamili K, et al. The relationship between alcohol consumption and chronic kidney disease in patients with nonalcoholic fatty liver disease. Scand J Gastroenterol 2024;Online ahead of print:1-9.
145. Wright WL, Urquhart S, Brunton S. Beyond blood glucose and blood pressure control in type 2 diabetes: alternative management strategies to prevent the development and progression of CKD. J Prim Care Community Health 2023;14:21501319231153599.
146. Brar S, Ye F, James MT, Harrison TG, Pannu N. Interdisciplinary Chronic Disease Collaboration (ICDC). Processes of care after hospital discharge for survivors of acute kidney injury: a population-based cohort study. Am J Kidney Dis 2024;83:216-28.
147. Maxson R, Starr J, Sewell J, Lyas C. SGLT2 inhibitors to slow chronic kidney disease progression: a review. Clin Ther 2024;46:e23-8.
148. Lee KC, Wu PS, Lin HC. Pathogenesis and treatment of non-alcoholic steatohepatitis and its fibrosis. Clin Mol Hepatol 2023;29:77-98.
149. Zhou H, Toshiyoshi M, Zhao W, Zhao Y, Zhao Y. Statins on nonalcoholic fatty liver disease: A systematic review and meta-analysis of 14 RCTs. Medicine 2023;102:e33981.
150. Jin Z, Yuan Y, Zheng C, Liu S, Weng H. Effects of sodium-glucose co-transporter 2 inhibitors on liver fibrosis in non-alcoholic fatty liver disease patients with type 2 diabetes mellitus: An updated meta-analysis of randomized controlled trials. J Diabetes Complications 2023;37:108558.
151. Athyros VG, Tziomalos K, Daskalopoulos GN, Karagiannis A, Mikhailidis DP. Statin-based treatment for cardiovascular risk and non-alcoholic fatty liver disease. Killing two birds with one stone? Ann Med 2011;43:167-71.
152. Spasovski G, Rroji M, Hristov G, Bushljetikj O, Spahia N, Rambabova Bushletikj I. A new hope on the horizon for kidney and cardiovascular protection with SGLT2 inhibitors, GLP-1 receptor agonists, and mineralocorticoid receptor antagonists in type 2 diabetic and chronic kidney disease patients. Metab Syndr Relat Disord 2024:Online ahead of print.
153. Sumida Y, Yoneda M, Toyoda H, et al. Common drug pipelines for the treatment of diabetic nephropathy and hepatopathy: can we kill two birds with one stone? Int J Mol Sci 2020;21:4939.
154. Tilg H, Byrne CD, Targher G. NASH drug treatment development: challenges and lessons. Lancet Gastroenterol Hepatol 2023;8:943-54.
155. Schnell O, Battelino T, Bergenstal R, et al. CVOT summit 2022 report: new cardiovascular, kidney, and glycemic outcomes. Cardiovasc Diabetol 2023;22:59.
156. Lin DS, Lee JK, Hung CS, Chen WJ. The efficacy and safety of novel classes of glucose-lowering drugs for cardiovascular outcomes: a network meta-analysis of randomised clinical trials. Diabetologia 2021;64:2676-86.
157. Banerjee M, Pal R, Maisnam I, Mukhopadhyay S. GLP-1 receptor agonists, SGLT2 inhibitors and noncardiovascular mortality in type 2 diabetes: Insights from a meta-analysis. Diabetes Metab Syndr 2024;18:102943.
158. Brockmeyer M, Parco C, Vargas KG, et al. Absolute treatment effects of novel antidiabetic drugs on a composite renal outcome: meta-analysis of digitalized individual patient data. J Nephrol 2024:Online ahead of print.
159. DeFronzo RA, Bakris GL. Modifying chronic kidney disease progression with the mineralocorticoid receptor antagonist finerenone in patients with type 2 diabetes. Diabetes Obes Metab 2022;24:1197-205.
160. Agarwal R, Filippatos G, Pitt B, et al. FIDELIO-DKD and FIGARO-DKD investigators. Cardiovascular and kidney outcomes with finerenone in patients with type 2 diabetes and chronic kidney disease: the FIDELITY pooled analysis. Eur Heart J 2022;43:474-84.
161. Adamson C, Jhund PS. Bringing FIDELITY to the estimate of treatment effects of finerenone in chronic kidney disease due to type 2 diabetes. Eur Heart J 2022;43:485-7.
162. Yamashita S, Masuda D, Matsuzawa Y. Clinical applications of a novel selective PPARα modulator, pemafibrate, in dyslipidemia and metabolic diseases. J Atheroscler Thromb 2019;26:389-402.
163. Horinouchi Y, Murashima Y, Yamada Y, et al. Pemafibrate inhibited renal dysfunction and fibrosis in a mouse model of adenine-induced chronic kidney disease. Life Sci 2023;321:121590.
164. Iwasaki M, Suzuki H, Umezawa Y, et al. Efficacy and safety of pemafibrate in patients with chronic kidney disease: A retrospective study. Medicine 2023;102:e32818.
165. Hadjivasilis A, Kouis P, Kousios A, Panayiotou A. The effect of fibrates on kidney function and chronic kidney disease progression: a systematic review and meta-analysis of randomised studies. J Clin Med 2022;11:768.
166. Yokote K, Yamashita S, Arai H, et al. Long-term efficacy and safety of pemafibrate, a novel selective peroxisome proliferator-activated receptor-α modulator (SPPARMα), in dyslipidemic patients with renal impairment. Int J Mol Sci 2019;20:706.
167. Goto H, Iseri K, Hida N. Fibrates and the risk of cardiovascular outcomes in chronic kidney disease patients. Nephrol Dial Transplant 2023;Online ahead of print:gfad248.
168. Kim DH, Park JS, Choi HI, et al. The critical role of FXR is associated with the regulation of autophagy and apoptosis in the progression of AKI to CKD. Cell Death Dis 2021;12:320.
169. Neuschwander-Tetri BA, Loomba R, Sanyal AJ, et al. NASH Clinical Research Network. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet 2015;385:956-65.
170. Ratziu V, Harrison SA, Loustaud-Ratti V, et al. Hepatic and renal improvements with FXR agonist vonafexor in individuals with suspected fibrotic NASH. J Hepatol 2023;78:479-92.
171. Dalbeni A, Garbin M, Zoncapè M, et al. Glomerular hyperfiltration: a marker of fibrosis severity in metabolic associated steatotic liver disease in an adult population. Int J Mol Sci 2023;24:15837.
172. Golabi P, Paik JM, Kumar A, et al. Nonalcoholic fatty liver disease (NAFLD) and associated mortality in individuals with type 2 diabetes, pre-diabetes, metabolically unhealthy, and metabolically healthy individuals in the United States. Metabolism 2023;146:155642.
173. Chung GE, Han K, Lee KN, et al. Combined effects of chronic kidney disease and nonalcoholic fatty liver disease on the risk of cardiovascular disease in patients with diabetes. Biomedicines 2022;10:1245.
174. Hydes TJ, Kennedy OJ, Buchanan R, et al. The impact of non-alcoholic fatty liver disease and liver fibrosis on adverse clinical outcomes and mortality in patients with chronic kidney disease: a prospective cohort study using the UK Biobank. BMC Med 2023;21:185.
175. Li Y, Wu S, Gao J, et al. Association of stroke with metabolic dysfunction-associated fatty liver disease with and without CKD. Am J Kidney Dis 2024;83:477-88.
176. Triozzi JL, Richardson PA, Gregg LP, Navaneethan SD. Incidence and predictors of non-alcoholic fatty liver disease among patients with chronic kidney disease. Nephrol Dial Transplant 2021;36:1546-8.
177. Wong F, Reddy KR, O'Leary JG, et al. Impact of chronic kidney disease on outcomes in cirrhosis. Liver Transpl 2019;25:870-80.
178. Nascimbeni F, Pellegrini E, Lugari S, et al. Statins and nonalcoholic fatty liver disease in the era of precision medicine: more friends than foes. Atherosclerosis 2019;284:66-74.
179. Ballestri S, Nascimbeni F, Romagnoli D, Baldelli E, Lonardo A. The role of nuclear receptors in the pathophysiology, natural course, and drug treatment of NAFLD in humans. Adv Ther 2016;33:291-319.
180. Kim HS, Lee J, Kim EH, et al. Association of myosteatosis with nonalcoholic fatty liver disease, severity, and liver fibrosis using visual muscular quality map in computed tomography. Diabetes Metab J 2023;47:104-17.
181. Arrese M, Cabello-verrugio C, Arab JP, et al. Sarcopenia in the setting of nonalcoholic fatty liver. Metab Target Organ Damage 2022;2:2.
182. Jankowski J, Floege J, Fliser D, Böhm M, Marx N. Cardiovascular disease in chronic kidney disease: pathophysiological insights and therapeutic options. Circulation 2021;143:1157-72.
183. Mace ML, Egstrand S, Morevati M, Olgaard K, Lewin E. New insights to the crosstalk between vascular and bone tissue in chronic kidney disease-mineral and bone disorder. Metabolites 2021;11:849.
184. Lonardo A. Renaming NAFLD to MAFLD: Could the LDE system assist in this transition? J Clin Med 2021;10:492.
185. Eslam M, Newsome PN, Sarin SK, et al. A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J Hepatol 2020;73:202-9.
186. Malhi H, Brown RS Jr, Lim JK, et al. Precipitous changes in nomenclature and definitions-NAFLD becomes SLD: implications for and expectations of AASLD journals. Hepatology 2023;78:1680-1.
187. Di Sessa A, Guarino S, Umano GR, Miraglia Del Giudice E, Marzuillo P. MASLD vs. NAFLD: A better definition for children with obesity at higher risk of kidney damage. J Hepatol 2024;80:e87-9.
188. Kobayashi N, Tada T, Nishimura T, et al. Metabolic dysfunction-associated steatotic liver disease criteria may underestimate the number of lean female nonalcoholic fatty liver disease patients with significant liver fibrosis. Hepatol Res 2023; doi: 10.1111/hepr.13994.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Lonardo A. Association of NAFLD/NASH, and MAFLD/MASLD with chronic kidney disease: an updated narrative review. Metab Target Organ Damage 2024;4:16. http://dx.doi.org/10.20517/mtod.2024.07
AMA Style
Lonardo A. Association of NAFLD/NASH, and MAFLD/MASLD with chronic kidney disease: an updated narrative review. Metabolism and Target Organ Damage. 2024; 4(2): 16. http://dx.doi.org/10.20517/mtod.2024.07
Chicago/Turabian Style
Lonardo, Amedeo. 2024. "Association of NAFLD/NASH, and MAFLD/MASLD with chronic kidney disease: an updated narrative review" Metabolism and Target Organ Damage. 4, no.2: 16. http://dx.doi.org/10.20517/mtod.2024.07
ACS Style
Lonardo, A. Association of NAFLD/NASH, and MAFLD/MASLD with chronic kidney disease: an updated narrative review. Metab Target Organ Damage. 2024, 4, 16. http://dx.doi.org/10.20517/mtod.2024.07
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 5 clicks
Cite This Article 5 clicks

 Like This Article 2
likes
Like This Article 2
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.